Photocatalysis
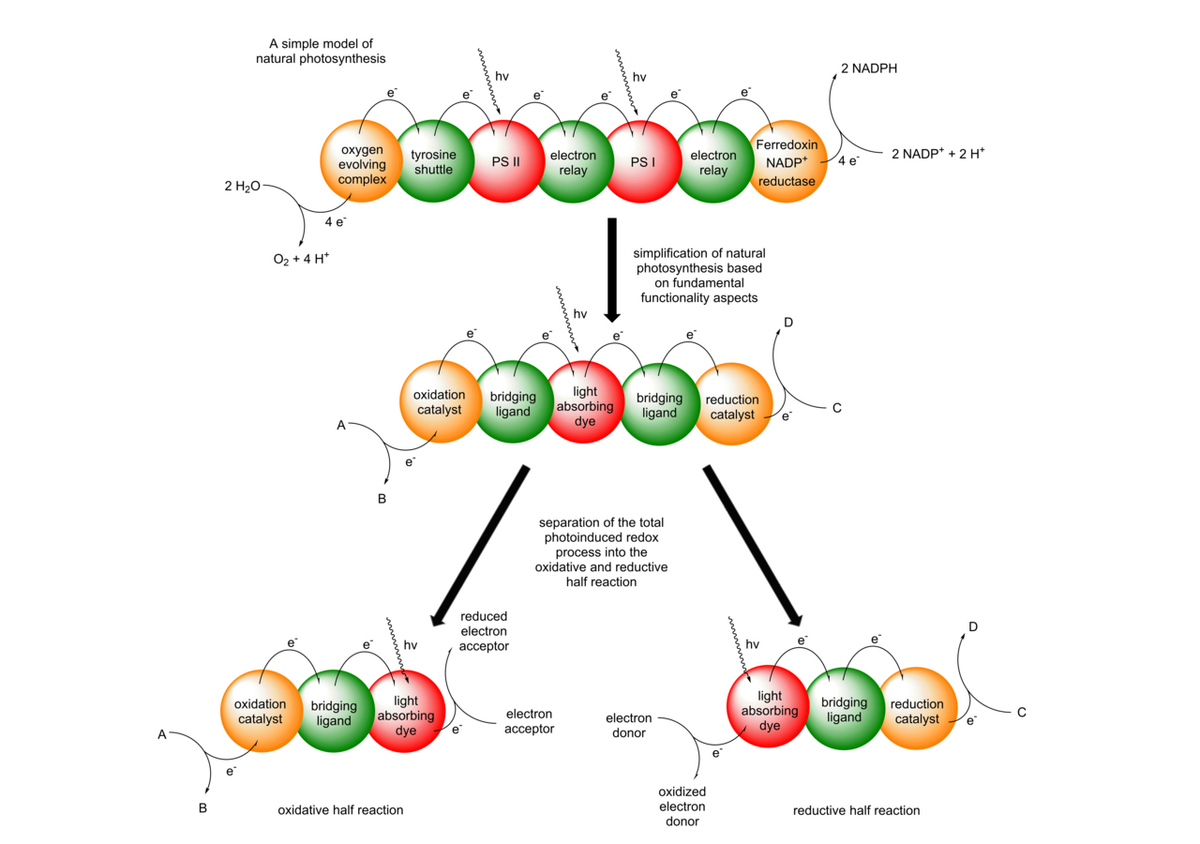
We are generally interested in designing, synthesizing and characterizing oligonuclear transition metal complexes for photoredox catalytic applications. Our goal is to understand the reaction mechanisms of our systems and to deviate structure property relationships, which allow for a knowledge-based optimization of our photocatalysts. Based on plants astonishing ability to use light for converting carbon dioxide into useful chemicals, we adapted some of nature´s key principles for the development of our molecular photocatalysts. A central research rationale in our group is hence the connection of visible light absorbing chromophores and catalytic centers via bridging ligands, which enables fast photoinduced vectorial electron transfers within the molecular scaffold. The outcome of this process is the generation of a charge separated state allowing redox reactions with suitable substrates to occur. As the complete water splitting reaction displays several challenges with respect to kinetic coupling of catalytic turnover and mechanistic insights, the oxidative and the reductive half reaction are primarily investigated separately within our group.
i) Photocatalytic reduction reactions
In the reductive half reaction, irradiation of the molecular assembly leads to an excitation of the Ru chromophore, followed by intramolecular electron transfers to the catalytic center. Hence, upon absorption of one photon, the Ru moiety is oxidized (RuII -> RuIII) and the catalytic center is reduced (Mn+ → M(n-1)+). To prevent the unproductive fast back electron transfer from the reduced catalytic center to the oxidized Ru center, sacrificial electron donors are added in excess, which can regenerate the initial RuII state. If this whole process occurs twice, the catalytic center becomes doubly reduced, which makes reductions of suitable substrates at the catalytic center possible to occur.
At the moment we are working on two different types of substrates. We either reduce protons to molecular hydrogen or we reduce nicotinamides, such as the naturally occurring NAD(P)+ to NAD(P)H. In the latter case we can connect the light driven nicotinamide reduction to light independent enzymatic transformations (photobiocatalysis). In the case of the reduction of protons to H2, we developed multiple dinuclear photocatalysts. The catalytic center is either connected via a N,N-chelating binding sphere (using the tetrapyridophenazine (tpphz) ligand) to the Ru chromophore or via a NHC-based (imidazophenanthroline (ip)) bridging ligand.
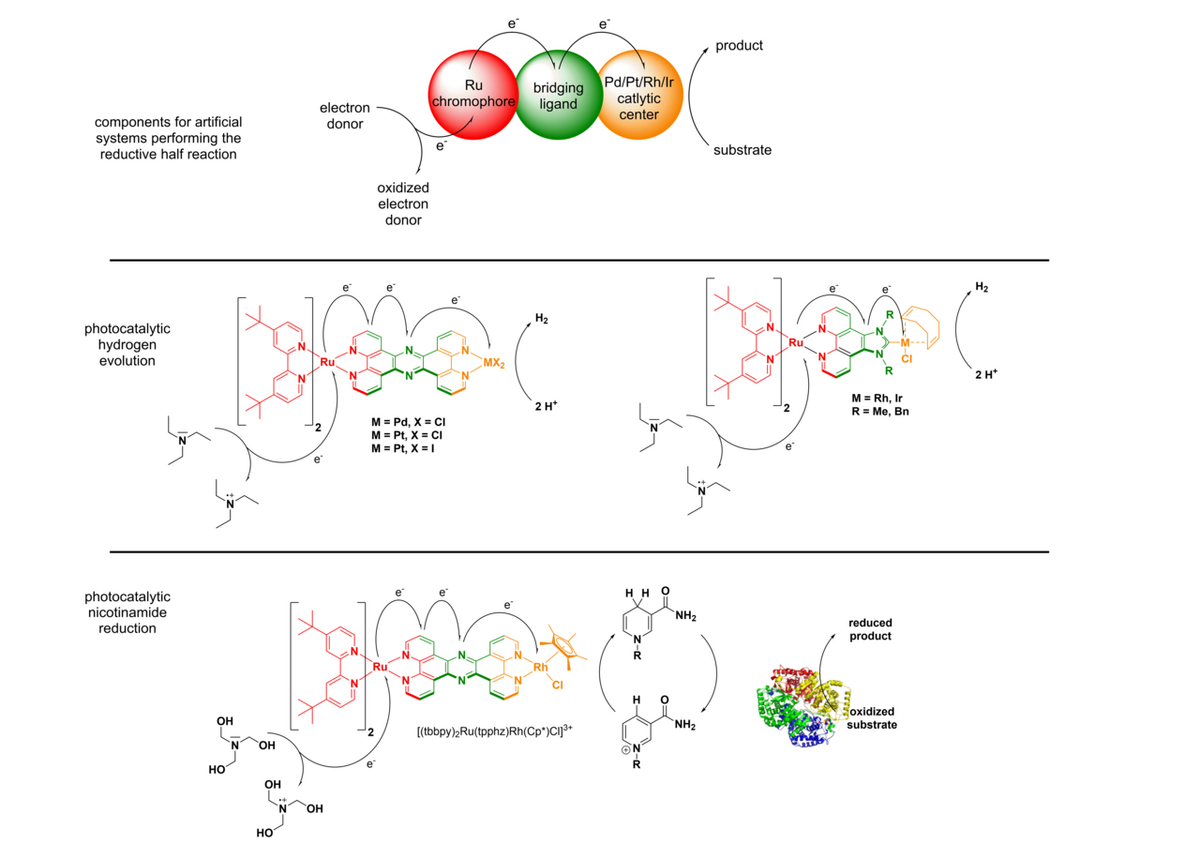
H2-Evolution with Ru(tpphz)-catalysts:
[1] S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry, J.G. Vos, Angew. Chem. Int. Ed. 2006, 45, 6215-6218.
[2] S. Tschierlei, M. Karnahl, M. Presselt, B. Dietzek, J. Guthmuller, L. Gonzalez, M. Schmitt, S. Rau, J. Popp, Angew. Chem. Int. Ed. 2010, 49, 3981-3984.
[3] M.G. Pfeffer, B. Schäfer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wächtler, B. Dietzek, V. Sundström, S. Rau, Angew. Chem. Int. Ed. 2015, 54, 5044-5048.
[4] M.G. Pfeffer, T. Kowacs, M. Wächtler, J. Guthmuller, B. Dietzek, J.G. Vos, S. Rau, Angew. Chem Int. Ed. 2015, 54, 6627-6631.
H2-Evolution with Ru(RRip)-catalysts:
[5] S. Kaufhold, L. Petermann, D. Sorsche, S. Rau, Chem. Eur. J. 2017, 23, 2271-2274.
[6] S. Kaufhold, D. Imanbaew, C. Riehn, S. Rau, Sustainable Energy Fuels 2017, 1, 2066-2070.
Photobiocatalysis:
[7] A. K. Mengele, G. M. Seibold, B. J. Eikmanns, S. Rau, ChemCatChem 2017, 9, 4369-4376.
ii) Photocatalytic Water-Oxidation
More recently our group took on the challenge of investigating the oxidative half-reaction of the overarching photocatalytic splitting of water. The oxidation of water into molecular oxygen and protons requires highly oxidizing conditions, four consecutive electron transfers and the formation of an O-O bond. Due to the harsh conditions and the complex nature of the reaction, photocatalytic water-oxidation is widely considered to be the bottleneck of artificial photosynthesis.
Because of this complexity there is only limited understanding of the underlying processes that govern catalyst reactivity and deactivation. This problem starts with detection of the evolved oxygen, since common analytical approaches (gas chromatography, electrochemical probes and pressure transducers) can often not be used in operando, investigate only one reaction phase (liquid or gas phase), consume oxygen and/or could lead to leakage from/into the reaction vessel as these techniques are invasive.
When faced with these challenges upon investigating literature known systems by Thummel et al. and Streb et al. our group was able to implement a measurement technique, which is non-invasive, does not require sampling and can be run in operando in both reaction phases. Here sensing is based on emission quenching of immobilized fluorescent dyes in the presence of molecular oxygen. These “sensor spots” can be mounted inside the reaction vessel and addressed from the outside.
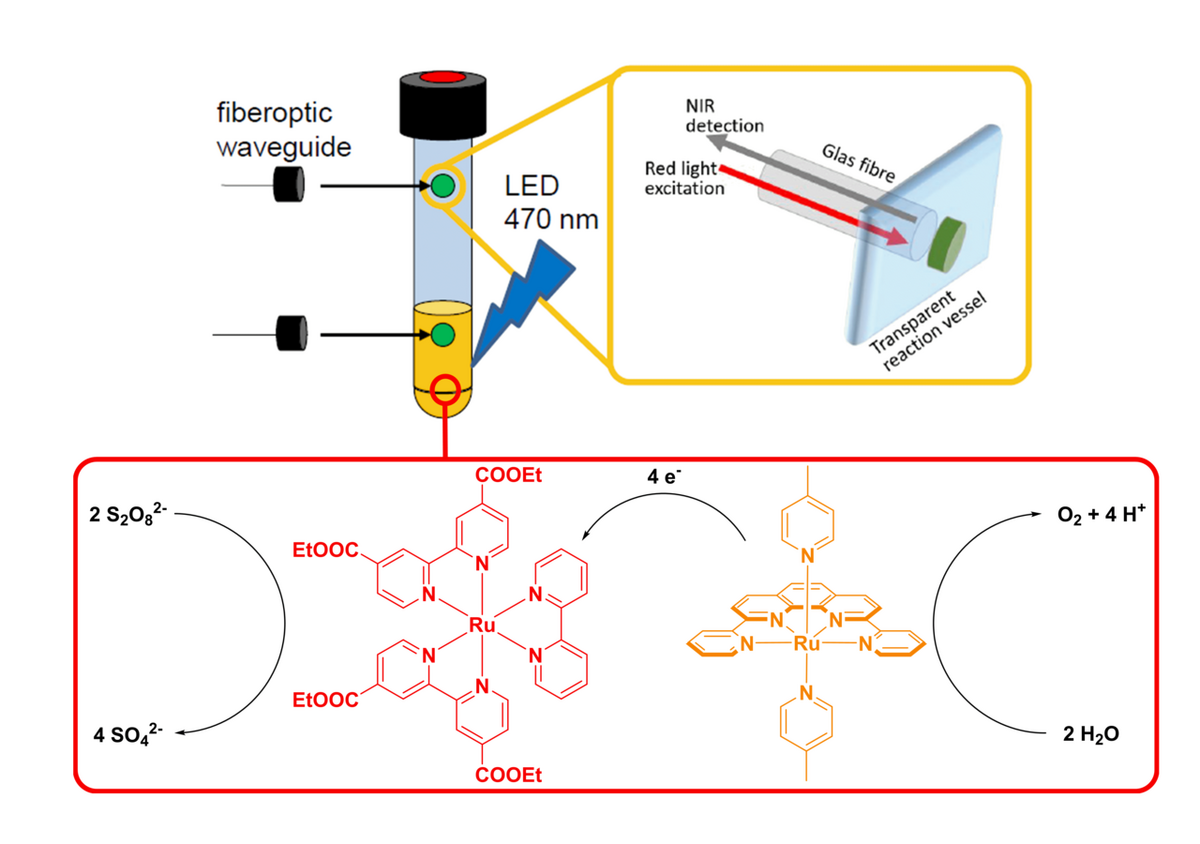
By utilizing this new approach to oxygen sensing we realized that the catalytic performance of some literature known systems was significantly underrated, additionally we were able to optimize systems and determine factors which hamper their performance in a quick and easy way. With this gained experience our group is currently working on the development, optimization and mechanistic investigation of new catalytic systems for the light driven oxidation of water.
In-operando measurement of O2:
[8] F. L. Huber, S. Amthor, B. Schwarz, B. Mizaikoff, C. Streb, S. Rau, Sustainable Energy Fuels 2018, 2, 1974-1978.