Central-Core und Multi-Minicore Myopathie
Bei beiden Krankheiten scheint auf Grund eigener Beobachtungen körperliches Ausdauertraining günstig zu sein.
Central-Core Myopathie
Angeborene Hypotrophie und Hypotonie der Muskulatur (floppy infant) und eine verzögerte motorische Entwicklung sind typisch für diese Patienten, die häufig schon bei Geburt eine Hüftluxation zeigen und später eine Skoliose entwickeln. Im Vordergrund steht eine Schwäche der Becken- und Oberschenkelmuskulatur. Üblicherweise besteht keine Einschränkung der bulbären und der Atemmuskulatur und auch keine externe Ophthalmoplegie. Die meisten Betroffenen werden mit den Jahren kräftiger und können selbständig gehen. Nur eine Minderheit der Kinder zeigt einen progredienten Verlauf. Die Abgrenzung zur Nemalin-Myopathie, zur zentronukleären Myopathie und zur Multi-Minicore Myopathie kann in Einzelfällen schwierig sein.
Die Erkrankung wird in der Mehrzahl der Fälle autosomal dominant mit wechselnder klinischer Ausprägung vererbt. Viele Familienangehörige eines Patienten weisen erst bei der genetischen Untersuchung die Anlage zur Myopathie auf. Da der Myopathie wie der Anlage zu maligner Hyperthermie RYR1-Mutationen zugrunde liegen, geht man davon aus, dass erkrankte und nicht erkrankte Mutationsträger in der Regel die Anlage zur MH tragen, auch wenn Ausnahmen beschrieben wurden. Diese Ausnahmen sind durch Mutationen im C-terminalen Bereich des Ryanodinrezeptors verursacht, die im Gegensatz zu den meisten anderen Mutationen nicht mit einer erhöhten, sondern mit einer verringerten Ca-Freisetzung einhergehen. Diese verminderte Freisetzung könnte die Muskelschwäche erklären. Aber auch Mutationen, die zu einer spontan erhöhten Öffnungswahrscheinlichkeit des Frei¬setzungskanals führen („leaky channel“), könnten über eine Depletion der sarkoplasma¬tischen Ca-Speicher die Schwäche bedingen.
Im Serum ist die CK meist normal oder nur leicht erhöht. In der Muskelbiopsie fällt die ausgeprägte Dominanz von Typ 1 Fasern auf. In der NADH-Färbung zeigen sich die charakteristischen, zentral gelegenen Cores, in denen die Mitochondrien zerstört sind. In Längsschnitten sieht man, dass sich ein Core meist über die gesamte Länge einer Typ 1 Faser erstreckt.
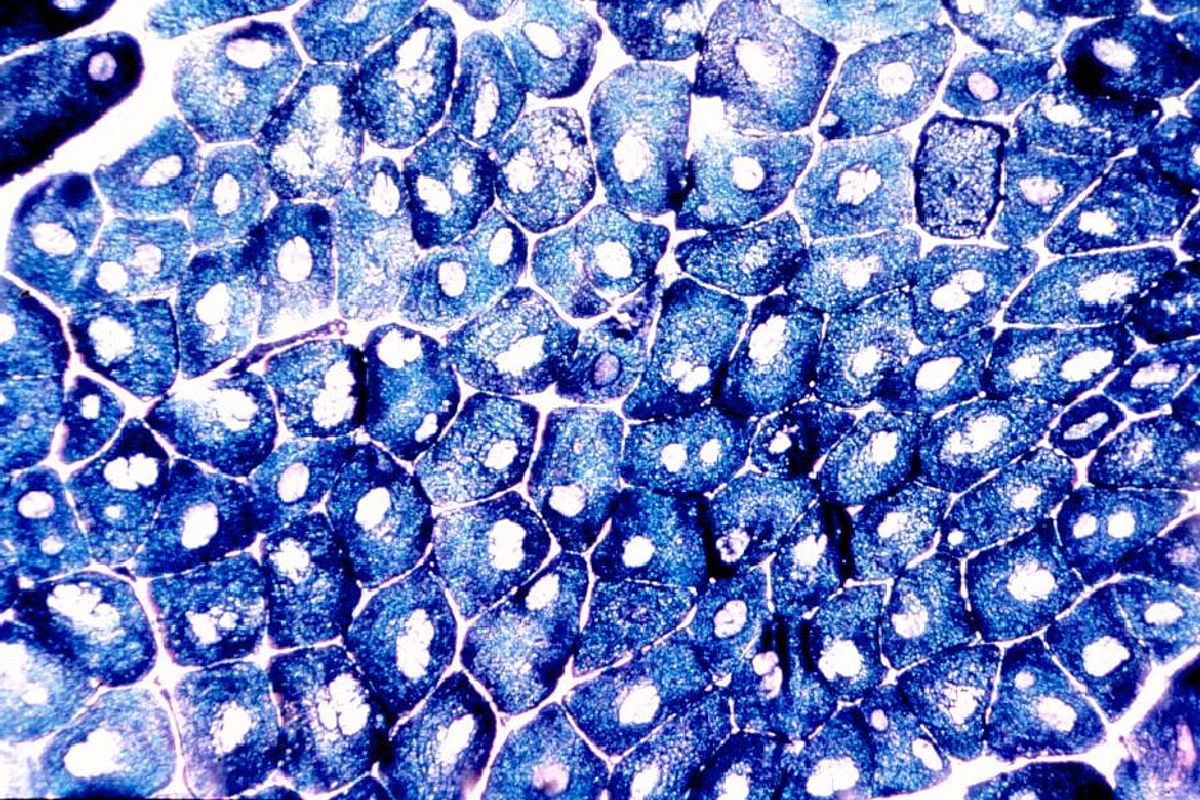
Multi-Minicore Myopathie
Die Multi-Minicore Myopathie ist eine rezessiv vererbte kongenitale Myopathie mit genetischer Heterogenität. Die klassische Form, die meist zu einer schweren Skoliose führt, zeigt eine achsiale Verteilung, d. h. respiratorische, bulbäre und extraokuläre Muskeln sind häufig betroffen. Diese Form wird durch Mutationen im SEPN1-Gen, das für Selenoprotein N ko¬diert, verursacht. Selenoprotein scheint für die Ca-Freisetzung aus dem sarkoplasmatischen Retikulum nötig zu sein.
Die Form, die durch RYR1-Mutationen verursacht wird, ist weniger schwer und eine Skoliose ist allenfalls schwach ausgeprägt. Es besteht eine generalisierte Muskelschwäche mit Betonung des Beckengürtels und der Handmuskeln mit Amyotrophie und Überstreckbarkeit der Gelenke. Wie bei der Central-Core Myopathie ist der Verlauf eher günstig und nur selten progredient. Auch die Histologie ist ähnlich. Allerdings dehenen sich die Cores nicht über die gesamte Faserlänge aus, sondern sind multipel und unscharf begrenzt. Mutationen in Patienten mit Core-Myopathien wurden auch im ACTA1-Gen für alpha-Aktin gefunden.
Die schwierige Frage der Vererbung mancher Core-Myopathien wird mit monoallelischer Expression erklärt. Diese genomische Prägung (imprinting) bezeichnet das Phänomen, dass die Genexpression davon abhängen kann, von welchem Elternteil das Allel stammt.