Publikationen 2017
Fast-NPS – A Markov Chain Monte Carlo- based analysis tool to obtain structural information from single-molecule FRET measurements

Tobias Eilert, Maximilian Beckers, Florian Drechsler, Jens Michaelis
The analysis tool and software package Fast-NPS can be used to analyse smFRET data to obtain quantitative structural information about macromolecules in their natural environment. In the algorithm a Bayesian model gives rise to a multivariate probability distribution describing the uncertainty of the structure determination. Since Fast-NPS aims to be an easy-to-use general-purpose analysis tool for a large variety of smFRET networks, we established an MCMC based sampling engine that approximates the target distribution and requires no parameter specification by the user at all. For an efficient local exploration we automatically adapt the multivariate proposal kernel according to the shape of the target distribution. In order to handle multimodality, the sampler is equipped with a parallel tempering scheme that is fully adaptive with respect to temperature spacing and number of chains. Since the molecular surrounding of a dye molecule affects its spatial mobility and thus the smFRET efficiency, we introduce dye models which can be selected for every dye molecule individually. These models allow the user to represent the smFRET network in great detail leading to an increased localisation precision. Finally, a tool to validate the chosen model combination is provided.
Computer Physics Communications
https://doi.org/10.1016/j.cpc.2017.05.027
DNA residence time is a regulatory factor of transcription repression
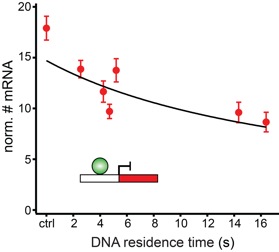
Karen Clauß Achim P. Popp Lena Schulze Johannes Hettich Matthias Reisser Laura Escoter Torres N. Henriette Uhlenhaut J. Christof M. Gebhardt
Transcription comprises a highly regulated sequence of intrinsically stochastic processes, resulting in bursts of transcription intermitted by quiescence. In transcription activation or repression, a transcription factor binds dynamically to DNA, with a residence time unique to each factor. Whether the DNA residence time is important in the transcription process is unclear. Here, we designed a series of transcription repressors differing in their DNA residence time by utilizing the modular DNA binding domain of transcription activator-like effectors (TALEs) and varying the number of nucleotide-recognizing repeat domains. We characterized the DNA residence times of our repressors in living cells using single molecule tracking. The residence times depended non-linearly on the number of repeat domains and differed by more than a factor of six. The factors provoked a residence time-dependent decrease in transcript level of the glucocorticoid receptor-activated gene SGK1. Down regulation of transcription was due to a lower burst frequency in the presence of long binding repressors and is in accordance with a model of competitive inhibition of endogenous activator binding. Our single molecule experiments reveal transcription factor DNA residence time as a regulatory factor controlling transcription repression and establish TALE-DNA binding domains as tools for the temporal dissection of transcription regulation.
Nucleic Acids Research
doi.org/10.1093/nar/gkx728
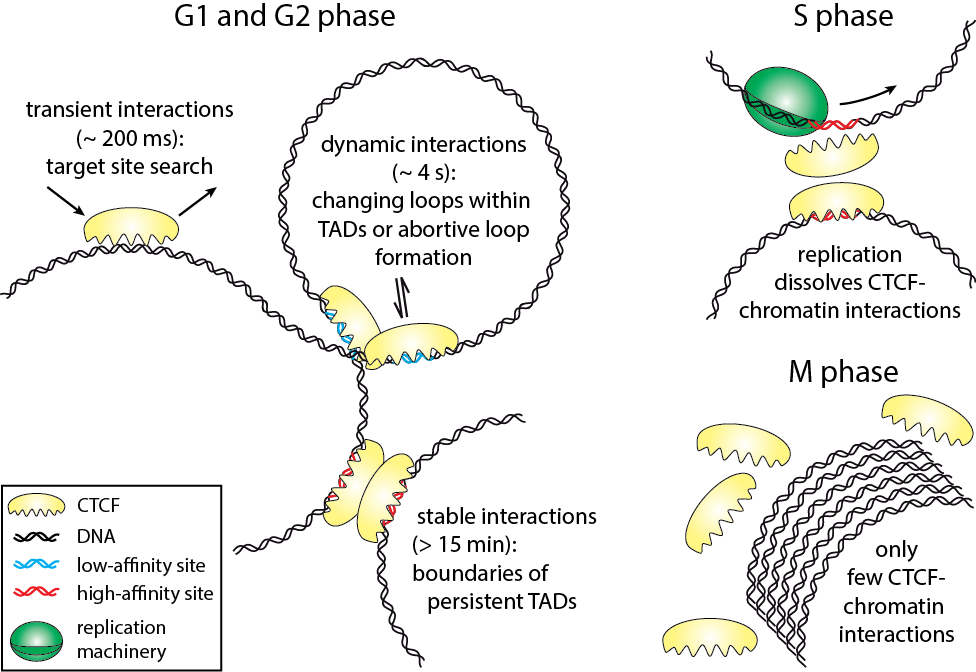
Direct Observation of Cell-Cycle-Dependent Interactions between CTCF and Chromatin
Harsha Agarwal, Matthias Reisser, Celina Wortmann, J. Christof M. Gebhardt
The three-dimensional arrangement of chromatin encodes regulatory traits important for nuclear processes such as transcription and replication. Chromatin topology is in part mediated by the architectural protein CCCTC-binding factor (CTCF) that binds to the boundaries of topologically associating domains. Whereas sites of CTCF interactions are well characterized, little is known on how long CTCF binds to chromatin and how binding evolves during the cell cycle. We monitored CTCF-chromatin interactions by live cell single molecule tracking in different phases of the cell cycle. In G1-, S-, and G2-phases, a majority of CTCF molecules was bound transiently (∼0.2 s) to chromatin, whereas minor fractions were bound dynamically (∼4 s) or stably (>15 min). During mitosis, CTCF was mostly excluded from chromatin. Our data suggest that CTCF scans DNA in search for two different subsets of specific target sites and provide information on the timescales over which topologically associating domains might be restructured. During S-phase, dynamic and stable interactions decreased considerably compared to G1-phase, but were resumed in G2-phase, indicating that specific interactions need to be dissolved for replication to proceed.
Biophysical Journal
doi.org/10.1016/j.bpj.2017.04.018
smFRET experiments of the RNA polymerase II transcription initiation complex
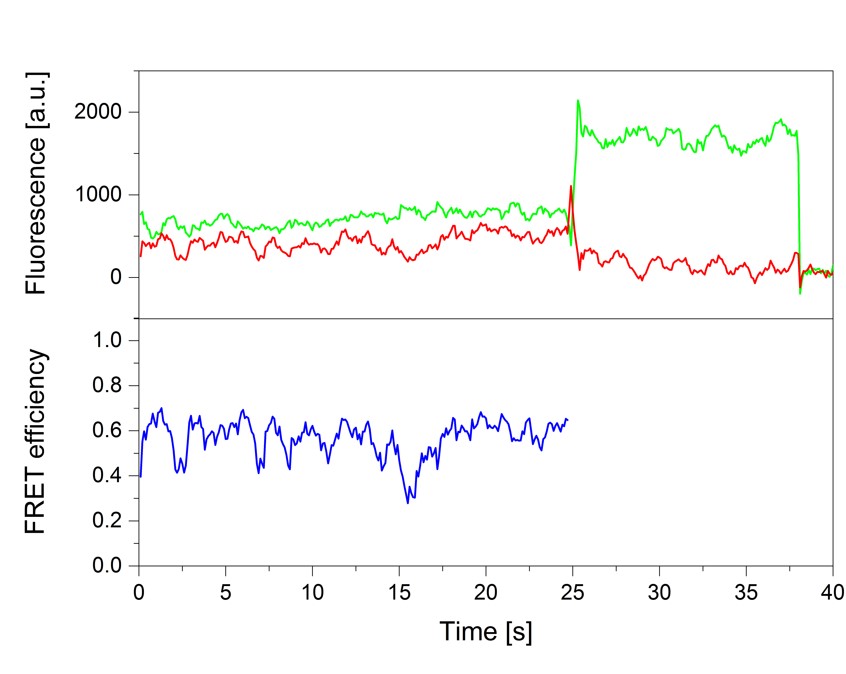
Nicole Malkusch, Thilo Dörfler, Julia Nagy, Tobias Eilert, Jens Michaelis
Single-molecule fluorescence and in particular single-molecule Förster Resonance Energy Transfer (smFRET) is a powerful tool to provide real-time information on the dynamic architecture of large macromolecular structures such as eukaryotic transcription initiation complexes. In contrast to other structural biology methods, not only structural details, but dynamics transitions are revealed thus closing in on the underlying molecular mechanisms. Here, we describe a comprehensive quantitative biophysical toolbox which can be used for rigorous analysis of dynamic protein-nucleic acid complexes and is applied to the study of eukaryotic transcription initiation. We present detailed protocols for the purification of all essential protein components of the minimal eukaryotic transcription initiation complex. Moreover, we demonstrate how elaborate strategies for site-specific protein labeling can be used to produce complexes with dye molecules attached to arbitrary desired positions. These complexes are then used for smFRET measurements. Moreover, we describe the Nano-Positioning System (NPS) which allows us to quantitatively use the results from a network of smFRET measurements to obtain structural information. With this we provide a toolbox to answer open questions which could not be addressed using methods like X-ray crystallography or cryo-electron microscopy.
Methods
doi.org/10.1016/j.ymeth.2017.04.011
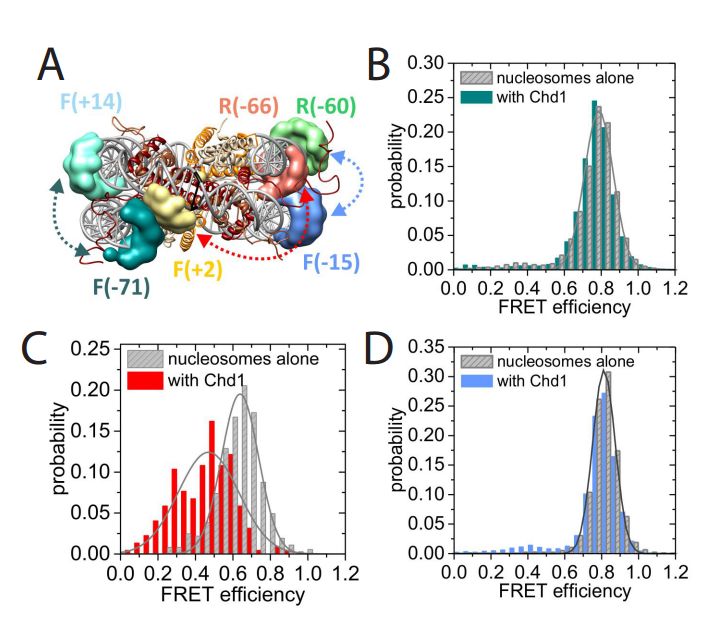
Structural reorganization of the chromatin remodeling enzyme Chd1 upon engagement with nucleosomes
Ramasubramanian Sundaramoorthy, Amanda L Hughes, Vijender Singh, Nicola Wiechens, Daniel P Ryan, Hassane El-Mkami, Maxim Petoukhov, Dmitri I svergun, Barbara Treutlein, Salina Quack, Monika Fischer, Jens Michaelis, Bettina Böttcher, David G Norman, Tom Owen-Hughes
The yeast Chd1 protein acts to position nucleosomes across genomes. Here we model the structure of the Chd1 protein in solution and when bound to nucleosomes. In the apo state the DNA binding domain contacts the edge of the nucleosome while in the presence of the non-hydrolyzable ATP analog, ADP-beryllium fluoride, we observe additional interactions between the ATPase domain and the adjacent DNA gyre 1.5 helical turns from the dyad axis of symmetry. Binding in this conformation involves unravelling the outer turn of nucleosomal DNA and requires substantial reorientation of the DNA binding domain with respect to the ATPase domains. The orientation of the DNA-binding domain is mediated by sequences in the N-terminus and mutations to this part of the protein have positive and negative effects on Chd1 activity. These observations indicate that the unfavourable alignment of C-terminal DNA binding region in solution contributes to an auto-inhibited state.
Cite as eLife
dx.doi.org/10.7554/eLife.22510
Atomic force microscopy of chromatin arrays reveal non-monotonic salt dependence of array compaction in solution

Katarzyna M. Krzemien, Maximilian Beckers, Salina Quack, Jens Michaelis
Compaction of DNA in chromatin is a hallmark of the eukaryotic cell and unravelling its structure is required for an understanding of DNA involving processes. Despite strong experimental efforts, many questions concerning the DNA packing are open. In particular, it is heavily debated whether an ordered structure referred to as the ª30 nm fibreº exist in vivo. Scanning probe microscopy has become a cutting edge technology for the high-resolution imaging of DNA- protein complexes. Here, we perform high-resolution atomic force microscopy of non-cross-linked chromatin arrays in liquid, under different salt conditions. A statistical analysis of the data reveals that array compaction is salt dependent in a non-monotonic fashion. A simple physical model can qualitatively explain the observed findings due to the opposing effects of salt dependent stiffening of DNA, nucleosome stability and histone-histone interactions. While for different salt concentrations different compaction states are observed, our data do not provide support for the existence of regular chromatin fibres. Our studies add new insights into chromatin structure, and with that contribute to a further understanding of the DNA condensation.
PLOS ONE
doi.org/10.1371/journal.pone.0173459
Structural Information from Single-molecule FRET Experiments Using the Fast Nano-positioning System
Thilo Dörfler, Tobias Eilert, Carlheinz Röcker, Julia Nagy, Jens Michaelis
Single-molecule Förster Resonance Energy Transfer (smFRET) can be used to obtain structural information on biomolecular complexes in realtime. Thereby, multiple smFRET measurements are used to localize an unknown dye position inside a protein complex by means of trilateration. In order to obtain quantitative information, the Nano-Positioning System (NPS) uses probabilistic data analysis to combine structural information from X-ray crystallography with single-molecule fluorescence data to calculate not only the most probable position but the complete threedimensional probability distribution, termed posterior, which indicates the experimental uncertainty. The concept was generalized for the analysis of smFRET networks containing numerous dye molecules. The latest version of NPS, Fast-NPS, features a new algorithm using Bayesian parameter estimation based on Markov Chain Monte Carlo sampling and parallel tempering that allows for the analysis of large smFRET networks in a comparably short time. Moreover, Fast-NPS allows the calculation of the posterior by choosing one of five different models for each dye, that account for the different spatial and orientational behavior exhibited by the dye molecules due to their local environment.
Journal of Visualized Experiments
doi:10.3791/54782