Dr. Sung Sakong
Ulm University
Institute of Theoretical Chemistry
Mez-Starck-Haus
Oberberghof 7
Institute of Theoretical Chemistry
Mez-Starck-Haus
Oberberghof 7
89081 Ulm
Germany
The theoretical study of vacuum/solid and liquid/solid interfaces requires a framework that can treat several material classes in different states of matter (solid, liquid, and/or gas-phase) reliably at the same time. Through quantum chemistry and density-functional-theory methods, we assess not only the surface/interface properties but also the microscopic processes and mechanisms of chemistry at interfaces. Our research interests in interfacial chemistry cover heterogeneous catalysis, electrocatalysis, physical chemistry, and electrochemistry.
Chemistry at surfaces and interfaces
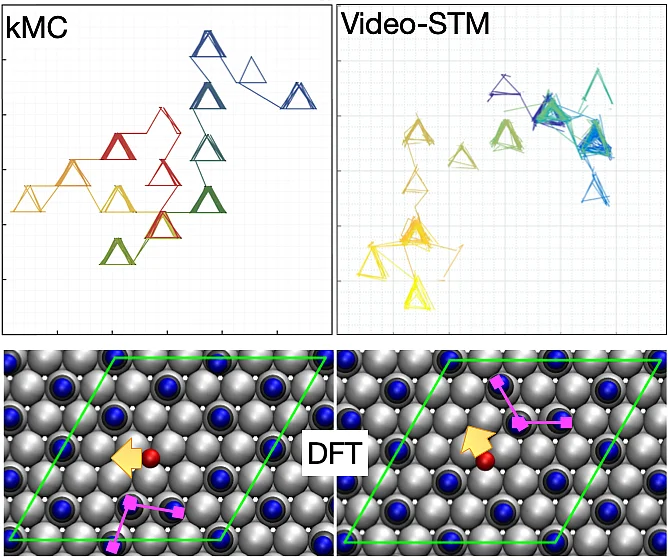
Using first-principles electronic structure methods, we explore rational catalyst design, search for improved electrode materials, and study the mechanism of photocatalysis. The elucidation of elementary surface and interface processes such as adsorption, desorption and diffusion of particles are still our primary interest. Based on potential energy surfaces derived from quantum chemistry calculations, additionally we employ kinetic Monte Carlo and molecular dynamics methods to investigate dynamical, statistical and kinetic properties of the studied systems.
Selected publications
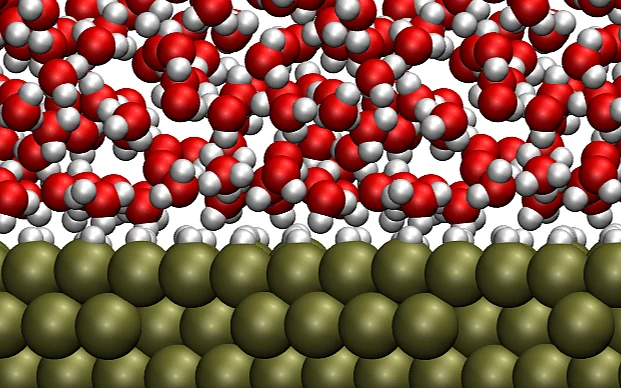
Modeling electric double layer
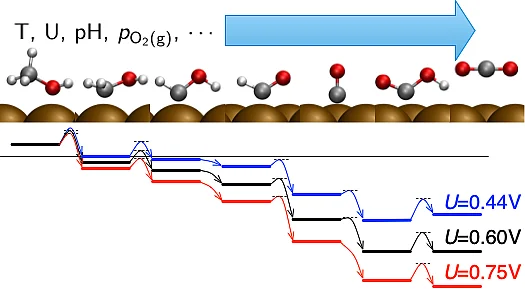
(Electro-)Catalytic reactions at surfaces and interfaces
Dynamics at surfaces