Publications 2022
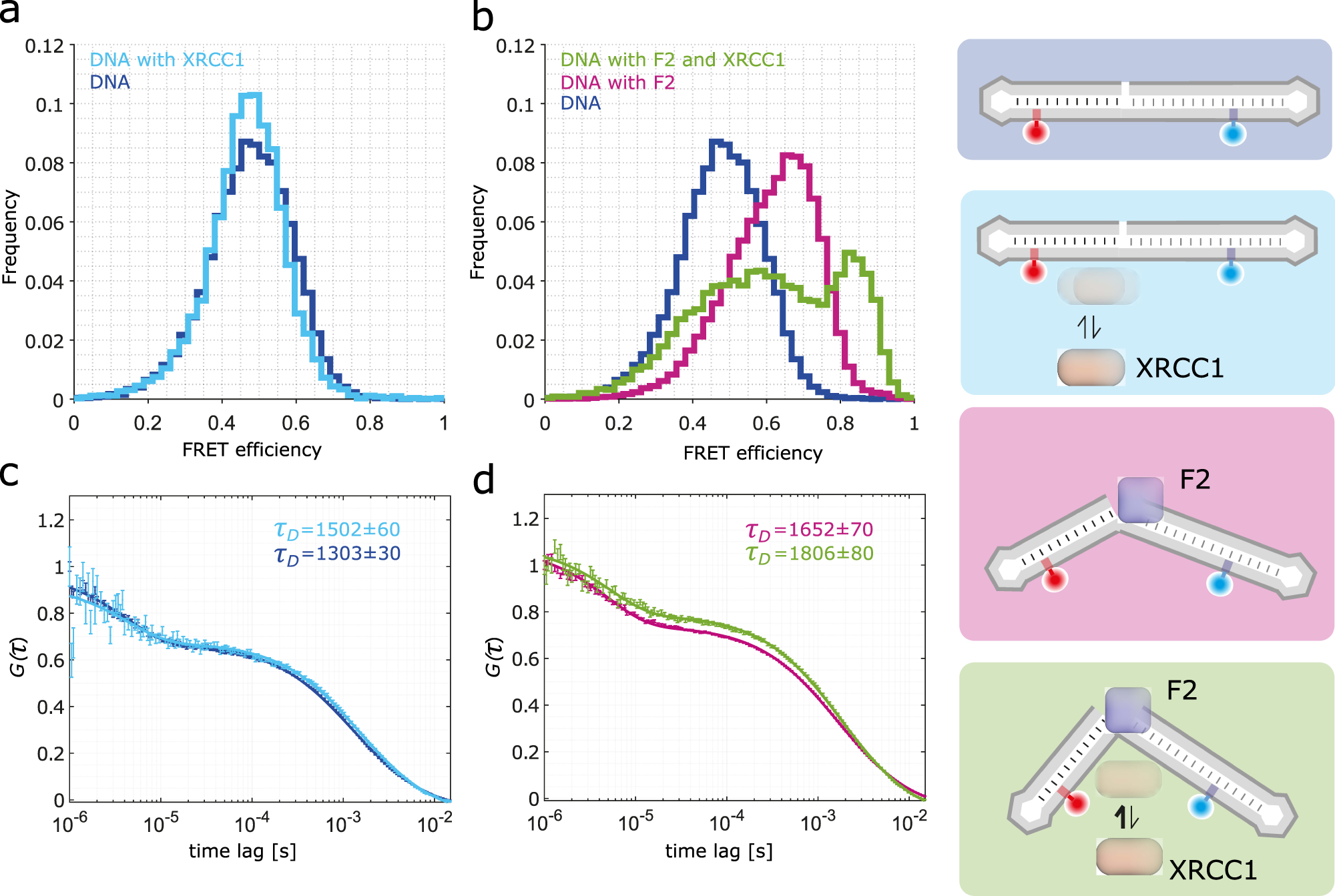
Structural dynamics of DNA strand break sensing by PARP-1 at a single-molecule level
Anna Sefer, Eleni Kallis, Tobias Eilert, Carlheinz Röcker, Olga Kolesnikova, David Neuhaus, Sebastian Eustermann, Jens Michaelis
Single-stranded breaks (SSBs) are the most frequent DNA lesions threatening genomic integrity. A highly kinked DNA structure in complex with human PARP-1 domains led to the proposal that SSB sensing in Eukaryotes relies on dynamics of both the broken DNA double helix and PARP-1’s multi-domain organization. Here, we directly probe this process at the single-molecule level. Quantitative smFRET and structural ensemble calculations reveal how PARP-1’s N-terminal zinc fingers convert DNA SSBs from a largely unperturbed conformation, via an intermediate state into the highly kinked DNA conformation. Our data suggest an induced fit mechanism via a multi-domain assembly cascade that drives SSB sensing and stimulates an interplay with the scaffold protein XRCC1 orchestrating subsequent DNA repair events. Interestingly, a clinically used PARP-1 inhibitor Niraparib shifts the equilibrium towards the unkinked DNA conformation, whereas the inhibitor EB47 stabilizes the kinked state.
https://www.nature.com/articles/s41467-022-34148-1
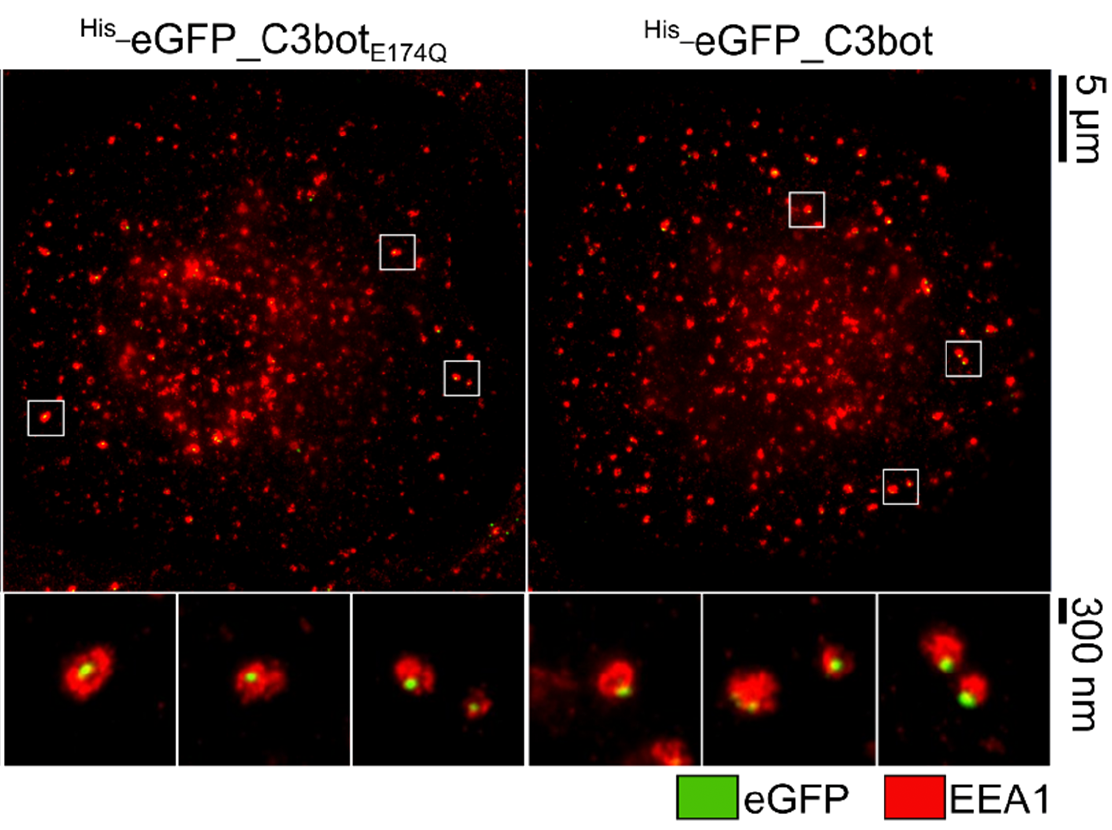
Clostridium botulinum C3 Toxin for Selective Delivery of Cargo into Dendritic Cells and Macrophages
Maximilian Fellermann, Mia Stemmer, Reiner Noschka, Fanny Wondany, Stephan Fischer, Jens Michaelis, Steffen Stenger, Holger Barth
The protein toxin C3bot from Clostridium botulinum is a mono-ADP-ribosyltransferase that selectively intoxicates monocyte-derived cells such as macrophages, osteoclasts, and dendritic cells (DCs) by cytosolic modification of Rho-A, -B, and -C. Here, we investigated the application of C3bot as well as its non-toxic variant C3botE174Q as transporters for selective delivery of cargo molecules into macrophages and DCs. C3bot and C3botE174Q facilitated the uptake of eGFP into early endosomes of human-monocyte-derived macrophages, as revealed by stimulated emission depletion (STED) super-resolution microscopy. The fusion of the cargo model peptide eGFP neither affected the cell-type selectivity (enhanced uptake into human macrophages ex vivo compared to lymphocytes) nor the cytosolic release of C3bot. Moreover, by cell fractionation, we demonstrated that C3bot and C3botE174Q strongly enhanced the cytosolic release of functional eGFP. Subsequently, a modular system was created on the basis of C3botE174Q for covalent linkage of cargos via thiol–maleimide click chemistry. The functionality of this system was proven by loading small molecule fluorophores or an established reporter enzyme and investigating the cellular uptake and cytosolic release of cargo. Taken together, non-toxic C3botE174Q is a promising candidate for the cell-type-selective delivery of small molecules, peptides, and proteins into the cytosol of macrophages and DCs.
https://www.mdpi.com/2072-6651/14/10/711
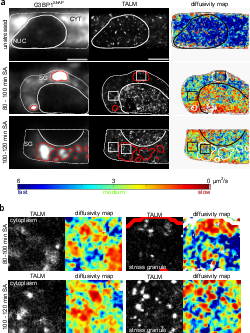
Stress induced TDP-43 mobility loss independent of stress granules
Lisa Streit, Timo Kuhn, Thomas Vomhof, Verena Bopp, Albert C. Ludolph, Jochen H. Weishaupt, Christof M. Gebhardt, Jens Michaelis & Karin M. Danzer
TAR DNA binding protein 43 (TDP-43) is closely related to the pathogenesis of amyotrophic lateral sclerosis (ALS) and translocates to stress granules (SGs). The role of SGs as aggregation-promoting “crucibles” for TDP-43, however, is still under debate. We analyzed TDP-43 mobility and localization under different stress and recovery conditions using live cell single-molecule tracking and super-resolution microscopy. Besides reduced mobility within SGs, a stress induced decrease of TDP-43 mobility in the cytoplasm and the nucleus was observed. Stress removal led to a recovery of TDP-43 mobility, which strongly depended on the stress duration. ‘Stimulated-emission depletion microscopy’ (STED) and ‘tracking and localization microscopy’ (TALM) revealed not only TDP-43 substructures within stress granules but also numerous patches of slow TDP-43 species throughout the cytoplasm. This work provides insights into the aggregation of TDP-43 in living cells and provide evidence suggesting that TDP-43 oligomerization and aggregation takes place in the cytoplasm separate from SGs.
https://doi.org/10.1038/s41467-022-32939-0
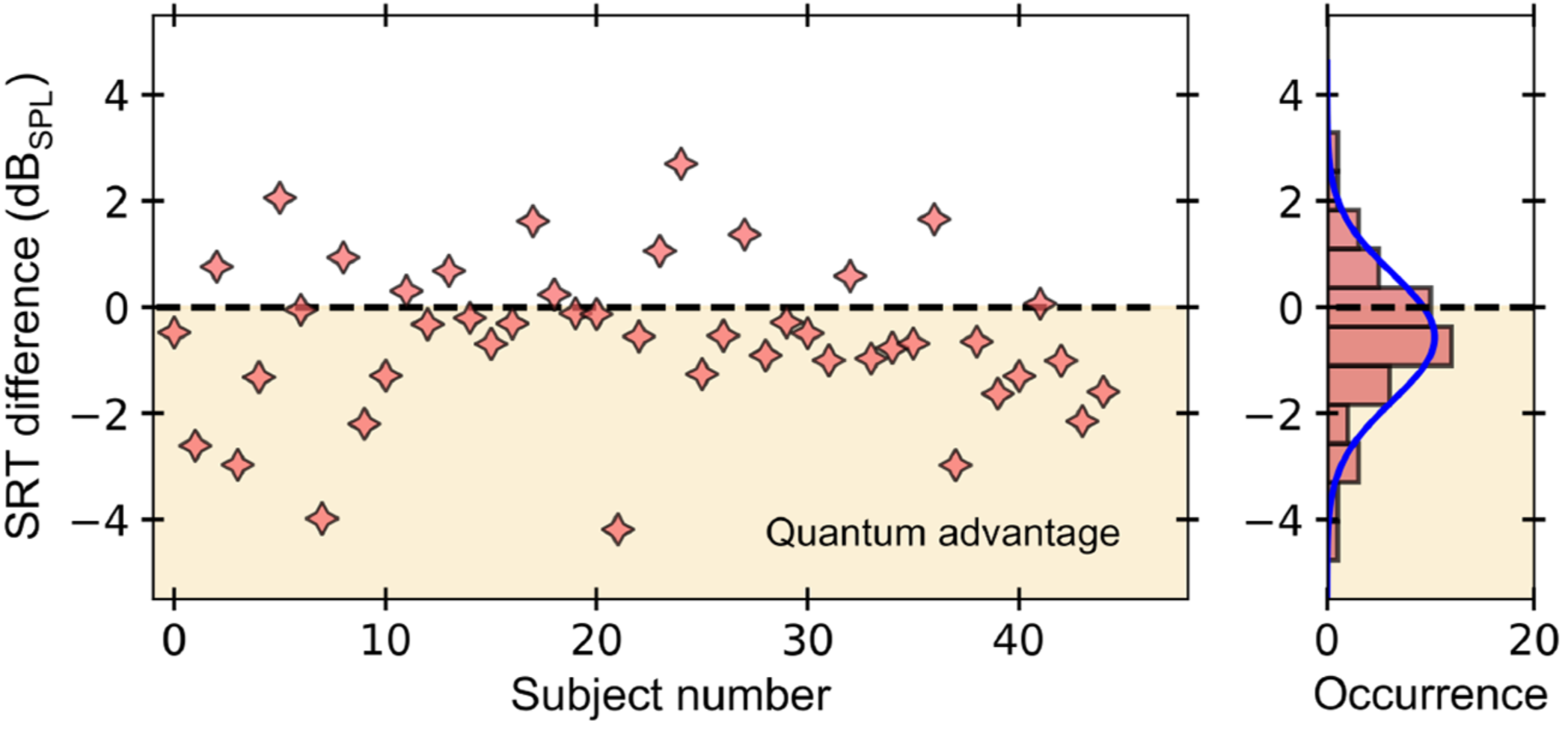
Quantum Optical Microphone in the Audio Band
Raphael Nold, Charles Babin, Joel Schmidt, Tobias Linkewitz, María T. Pérez Zaballos, Rainer Stöhr, Roman Kolesov, Vadim Vorobyov, Daniil M. Lukin, Rüdiger Boppert, Stefanie Barz, Jelena Vučković, J. Christof M. Gebhardt, Florian Kaiser, and Jörg Wrachtrup
The ability to perform high-precision optical measurements is paramount in science and engineering. Laser interferometry enables interaction-free sensing with a precision ultimately limited by shot noise. Quantum optical sensors can surpass this limit but single- or multiphoton schemes are challenged by low experimental sampling rates, while squeezed-light approaches require complex optical setups and sophisticated time gating. Here, we introduce a simple method that infers optical phase shifts through standard intensity measurements while still maintaining the quantum advantage in the measurement precision. Capitalizing on the robustness and high sampling rates of our device, we implement a quantum optical microphone in the audio band. Its performance is benchmarked against a classical laser microphone in a standardized medically approved speech-recognition test on 45 subjects. We find that quantum-recorded words improve the speech-recognition threshold by −0.57dBSPL, thus making the quantum advantage audible. Not only do these results open the door toward applications in quantum nonlinear interferometry but they also show that quantum phenomena can be experienced by humans.
https://doi.org/10.1103/PRXQuantum.3.020358
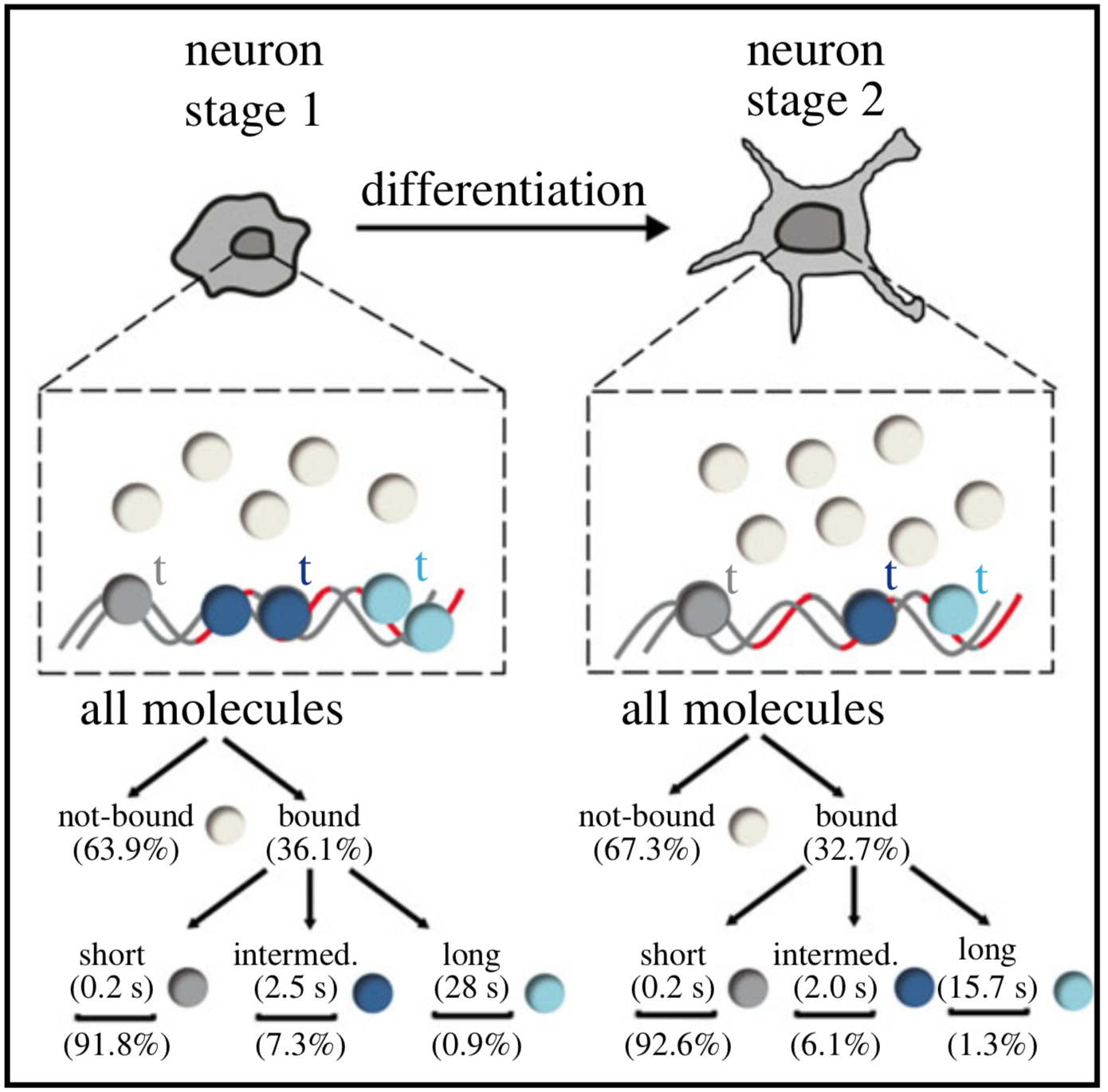
Single-molecule tracking (SMT) and localization of SRF and MRTF transcription factors during neuronal stimulation and differentiation
Oliver Kuchler, Jule Gerlach, Thomas Vomhof, Johannes Hettich, Julia Steinmetz, J. Christof M. Gebhardt, Jens Michaelis and Bernd Knöll
In cells, proteins encoded by the same gene do not all behave uniformly but engage in functional subpopulations induced by spatial or temporal segregation. While conventional microscopy has limitations in revealing such spatial and temporal diversity, single-molecule tracking (SMT) microscopy circumvented this problem and allows for high-resolution imaging and quantification of dynamic single-molecule properties. Particularly in the nucleus, SMT has identified specific DNA residence times of transcription factors (TFs), DNA-bound TF fractions and positions of transcriptional hot-spots upon cell stimulation. By contrast to cell stimulation, SMT has not been employed to follow dynamic TF changes along stages of cell differentiation. Herein, we analysed the serum response factor (SRF), a TF involved in the differentiation of many cell types to study nuclear single-molecule dynamics in neuronal differentiation. Our data in living mouse hippocampal neurons show dynamic changes in SRF DNA residence time and SRF DNA-bound fraction between the stages of adhesion, neurite growth and neurite differentiation in axon and dendrites. Using TALM (tracking and localization microscopy), we identified nuclear positions of SRF clusters and observed changes in their numbers and size during differentiation. Furthermore, we show that the SRF cofactor MRTF-A (myocardin-related TF or MKL1) responds to cell activation by enhancing the long-bound DNA fraction. Finally, a first SMT colocalization study of two proteins was performed in living cells showing enhanced SRF/MRTF-A colocalization upon stimulation. In summary, SMT revealed modulation of dynamic TF properties during cell stimulation and differentiation.
https://doi.org/10.1098/rsob.210383
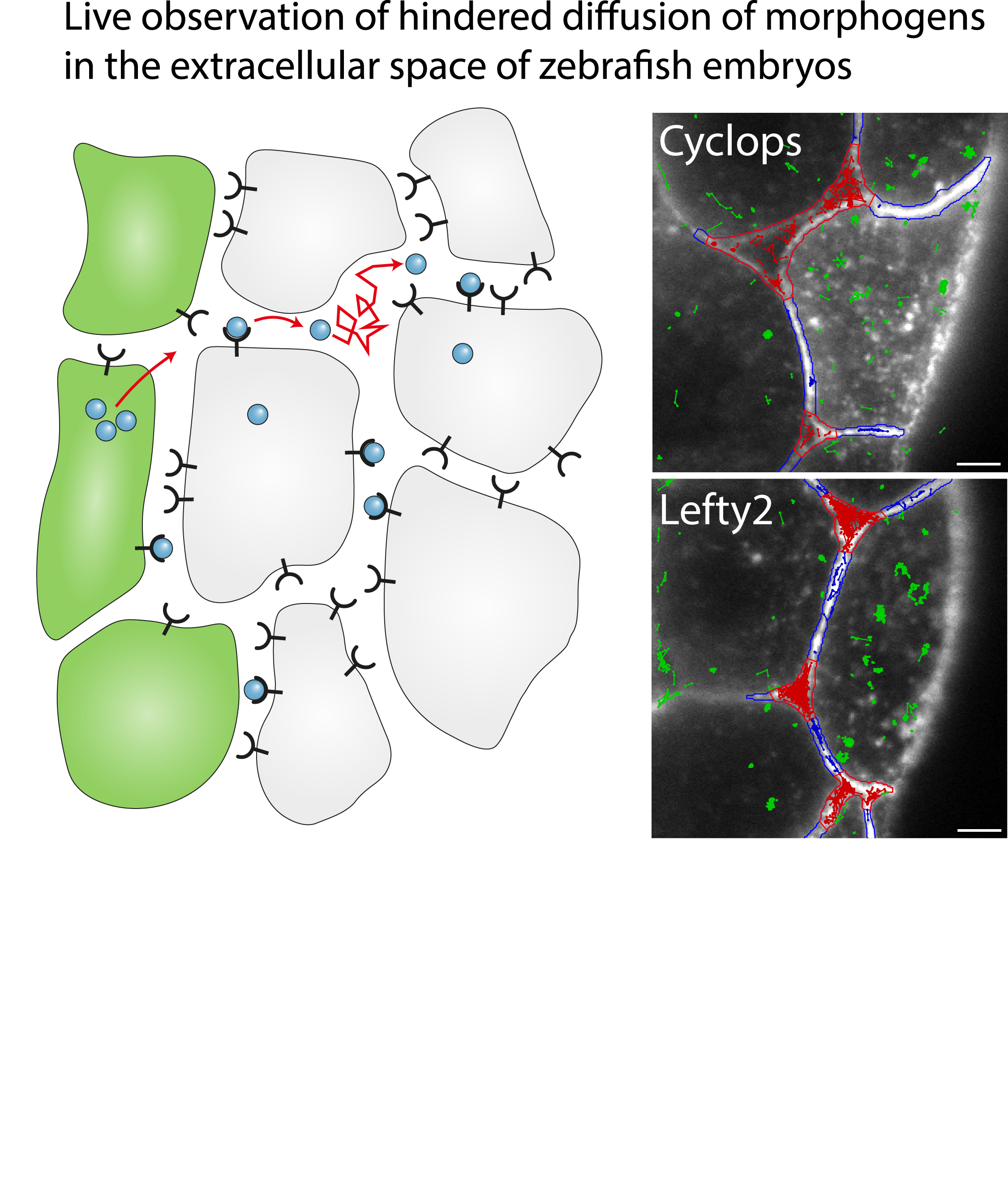
Single-molecule tracking of Nodal and Lefty in live zebrafish embryos supports hindered diffusion model
Timo Kuhn, Amit N. Landge, David Mörsdorf, Jonas Coßmann, Johanna Gerstenecker, Daniel Čapek, Patrick Müller and J. Christof M. Gebhardt
The hindered diffusion model postulates that the movement of a signaling molecule through an embryo is affected by tissue geometry and binding-mediated hindrance, but these effects have not been directly demonstrated in vivo. Here, we visualize extracellular movement and binding of individual molecules of the activator-inhibitor signaling pair Nodal and Lefty in live developing zebrafish embryos using reflected light-sheet microscopy. We observe that diffusion coefficients of molecules are high in extracellular cavities, whereas mobility is reduced and bound fractions are high within cell-cell interfaces. Counterintuitively, molecules nevertheless accumulate in cavities, which we attribute to the geometry of the extracellular space by agent-based simulations. We further find that Nodal has a larger bound fraction than Lefty and shows a binding time of tens of seconds. Together, our measurements and simulations provide direct support for the hindered diffusion model and yield insights into the nanometer-to-micrometer-scale mechanisms that lead to macroscopic signal dispersal.
https://doi.org/10.1038/s41467-022-33704-z
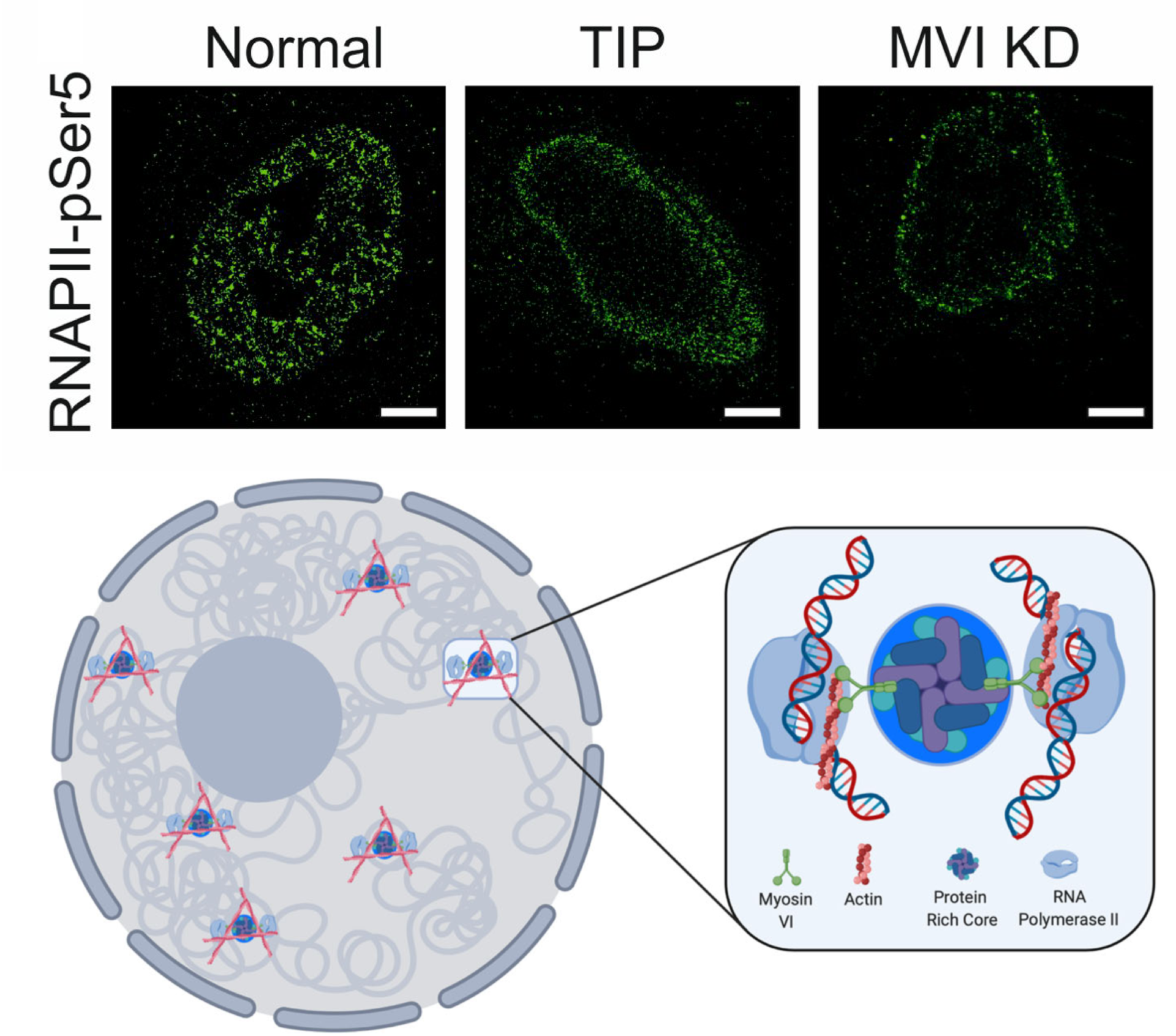
Myosin VI regulates the spatial organisation of mammalian transcription initiation
Yukti Hari-Gupta , Natalia Fili, Ália dos Santos, Alexander W. Cook, Rosemarie E. Gough,
Hannah C. W. Reed, Lin Wang, Jesse Aaron, Tomas Venit, Eric Wait,
Andreas Grosse-Berkenbusch, J. Christof M. Gebhardt, Piergiorgio Percipalle, Teng-Leong Chew,
Marisa Martin-Fernandez & Christopher P. Toseland
During transcription, RNA Polymerase II (RNAPII) is spatially organised within the nucleus into clusters that correlate with transcription activity. While this is a hallmark of genome regulation in mammalian cells, the mechanisms concerning the assembly, organisation and stability remain unknown. Here, we have used combination of single molecule imaging and genomic approaches to explore the role of nuclear myosin VI (MVI) in the nanoscale organisation of RNAPII. We reveal that MVI in the nucleus acts as the molecular anchor that holds RNAPII in high density clusters. Perturbation of MVI leads to the disruption of RNAPII localisation, chromatin organisation and subsequently a decrease in gene expression. Overall, we uncover the fundamental role of MVI in the spatial regulation of gene expression.
https://doi.org/10.1038/s41467-022-28962-w
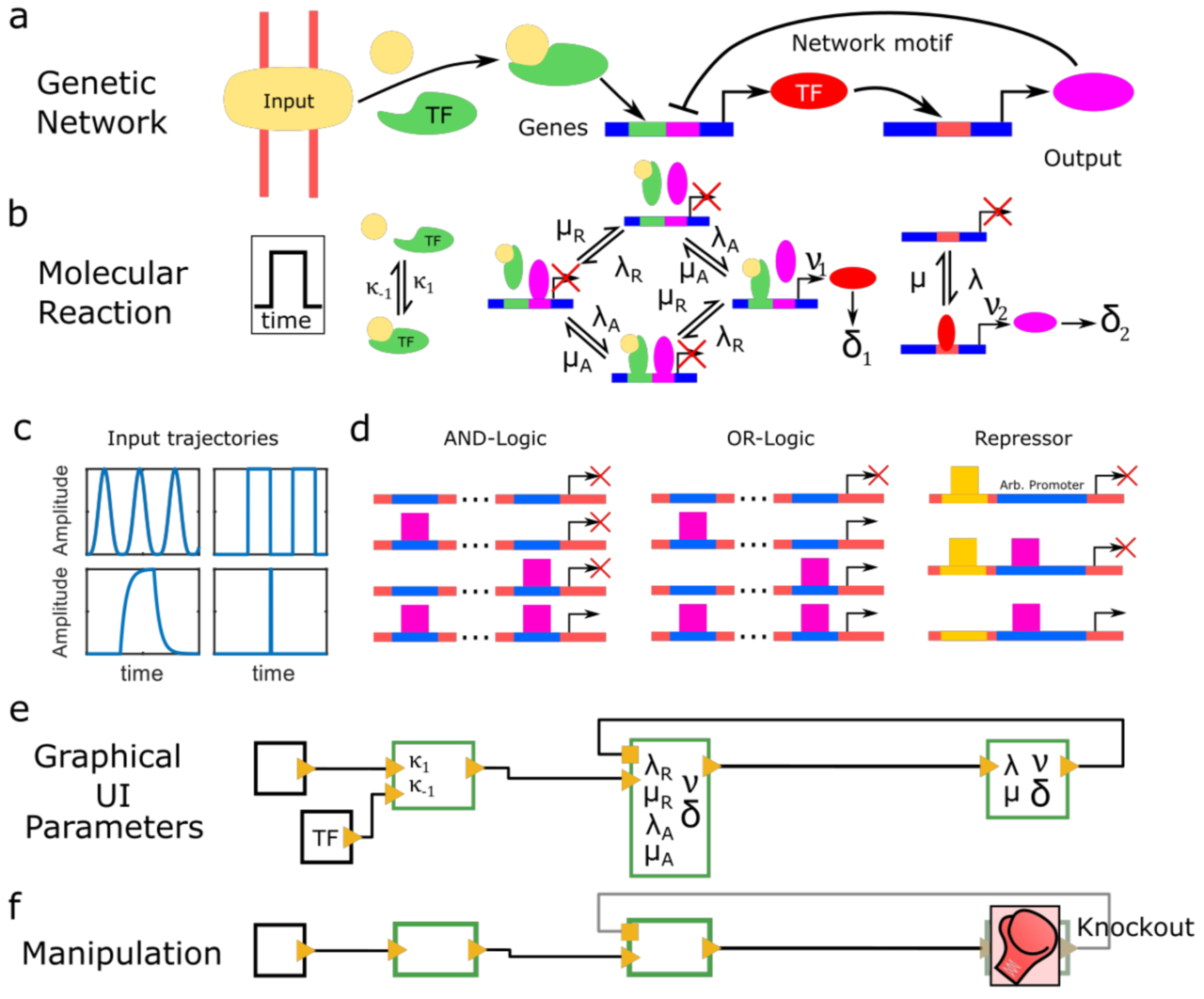
Periodic synchronization of isolated network elements facilitates simulating and inferring gene regulatory networks including stochastic molecular kinetics
Johannes Hettich and Christof M. Gebhardt
The temporal progression of many fundamental processes in cells and organisms, including homeostasis, differentiation and development, are governed by gene regulatory networks (GRNs). GRNs balance fluctuations in the output of their genes, which trace back to the stochasticity of molecular interactions. Although highly desirable to understand life processes, predicting the temporal progression of gene products within a GRN is challenging when considering stochastic events such as transcription factor–DNA interactions or protein production and degradation.
We report a method to simulate and infer GRNs including genes and biochemical reactions at molecular detail. In our approach, we consider each network element to be isolated from other elements during small time intervals, after which we synchronize molecule numbers across all network elements. Thereby, the temporal behaviour of network elements is decoupled and can be treated by local stochastic or deterministic solutions. We demonstrate the working principle of this modular approach with a repressive gene cascade comprising four genes. By considering a deterministic time evolution within each time interval for all elements, our method approaches the solution of the system of deterministic differential equations associated with the GRN. By allowing genes to stochastically switch between on and off states or by considering stochastic production of gene outputs, we are able to include increasing levels of stochastic detail and approximate the solution of a Gillespie simulation. Thereby, CaiNet is able to reproduce noise-induced bi-stability and oscillations in dynamically complex GRNs. Notably, our modular approach further allows for a simple consideration of deterministic delays. We further infer relevant regulatory connections and steady-state parameters of a GRN of up to ten genes from steady-state measurements by identifying each gene of the network with a single perceptron in an artificial neuronal network and using a gradient decent method originally designed to train recurrent neural networks. To facilitate setting up GRNs and using our simulation and inference method, we provide a fast computer-aided interactive network simulation environment, CaiNet.
We developed a method to simulate GRNs at molecular detail and to infer the topology and steady-state parameters of GRNs. Our method and associated user-friendly framework CaiNet should prove helpful to analyze or predict the temporal progression of reaction networks or GRNs in cellular and organismic biology.
https://doi.org/10.1186/s12859-021-04541-6