Publications 2023
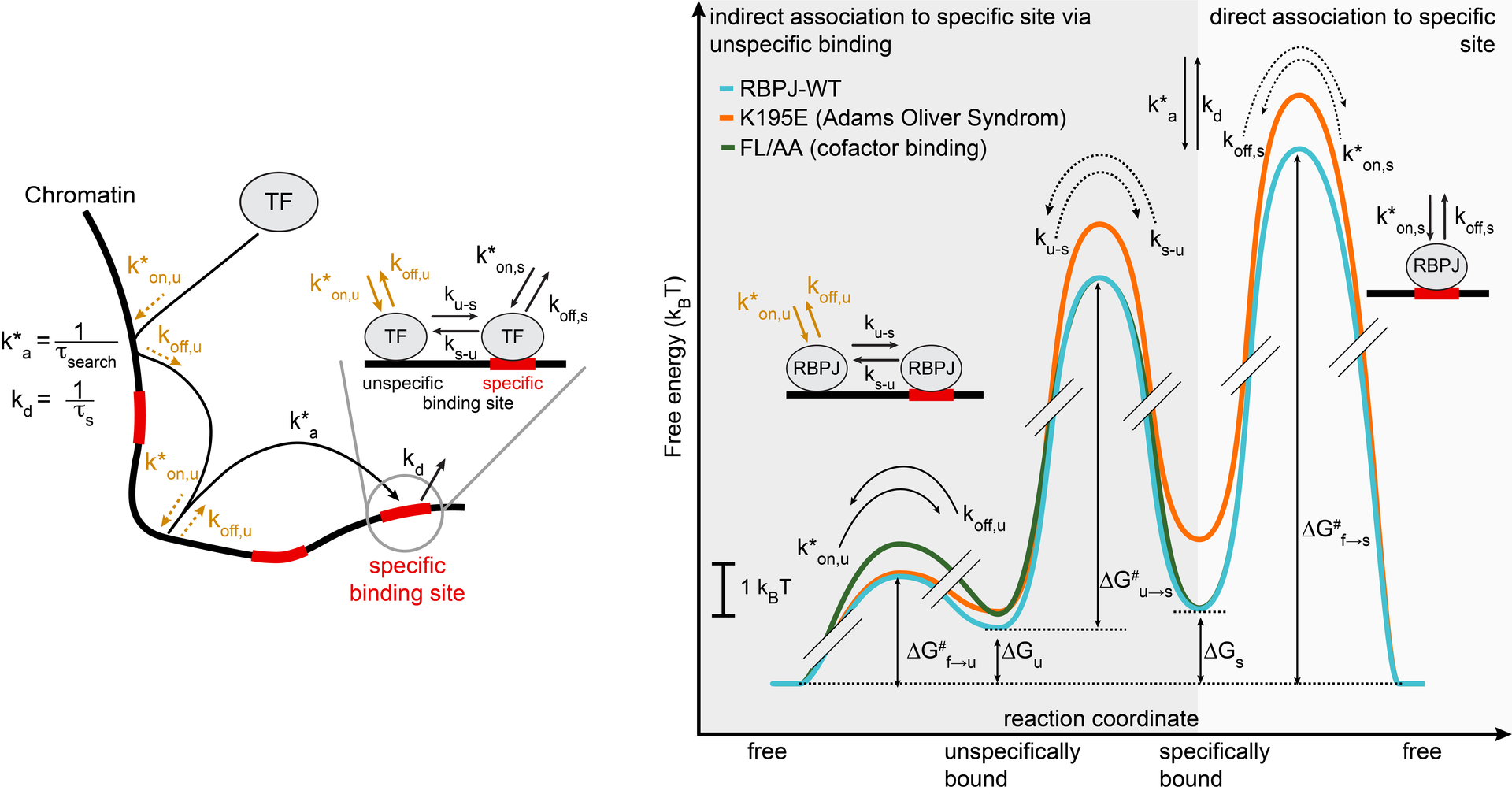
In vivo binding free energy landscape reveals kinetic control of transcription factor function
Duyen Huynh, Philipp Hoffmeister, Tobias Friedrich, Kefan Zhang, Marek Bartkuhn, Francesca Ferrante, Benedetto Daniele Giaimo, Rhett Kovall, Tilman Borggrefe, Franz Oswald, J. Christof M. Gebhardt
Transcription factors such as RBPJ in Notch signal transduction bind to specific DNA sequences and initiate either repression or activation of genes. Which sites they select and how often and long they bind affects the efficiency of gene regulation. To resolve the underpinnings of RBPJ-DNA binding, we determined the in vivo binding free energy landscape of RBPJ using live-cell single-molecule tracking and genome-wide chromatin immunoprecipitation. Importantly, DNA binding of RBPJ was thermodynamically unstable in vivo and instead governed by the binding kinetics: Cofactors contributed to target site specificity by tuning both association and dissociation of unspecific binding, while mutation K195E underlying Adams-Oliver-Syndrome destabilized specific DNA binding by mainly altering the association rate. We showed thermodynamic instability in vivo also for other transcription factors, indicating that kinetic rather than thermodynamic control of DNA binding might be a general feature of transcription factors in vivo.
www.biorxiv.org/content/10.1101/2023.12.19.572376v1
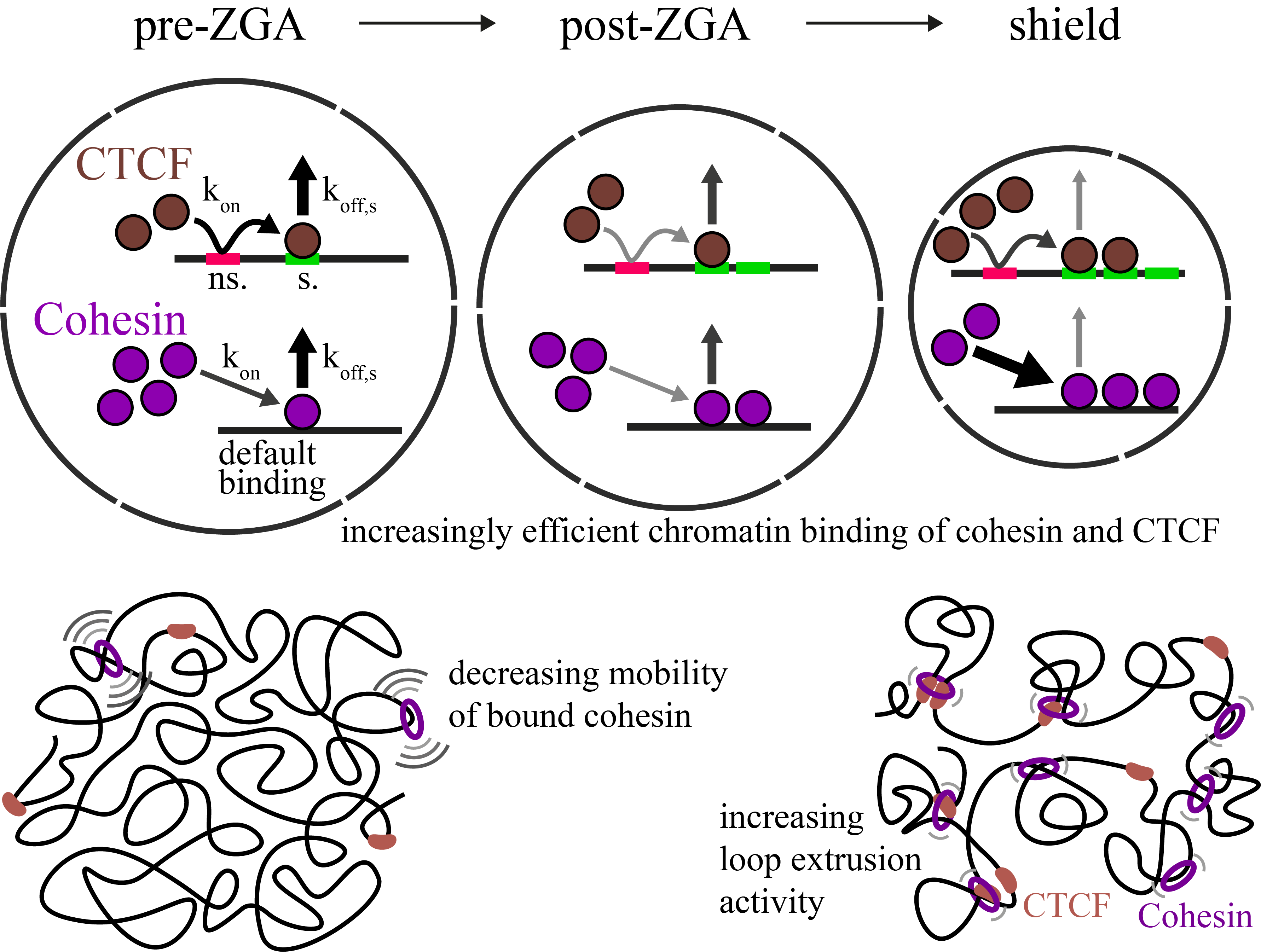
Increasingly efficient chromatin binding of cohesin and CTCF supports chromatin architecture formation during zebrafish embryogenesis
The three-dimensional folding of chromosomes is essential for nuclear functions such as DNA replication and gene regulation. The emergence of chromatin architecture is thus an important process during embryogenesis. To shed light on the molecular and kinetic underpinnings of chromatin architecture formation, we characterized biophysical properties of cohesin and CTCF binding to chromatin and their changes upon cofactor depletion using single-molecule imaging in live developing zebrafish embryos. We found that chromatin-bound fractions of both cohesin and CTCF increased significantly between the 1000-cell and shield stages, which we could explain through changes in both their association and dissociation rates. Moreover, increasing binding of cohesin restricted chromatin motion, potentially via loop extrusion, and showed distinct stage-dependent nuclear distribution. Polymer simulations with experimentally derived parameters recapitulated the experimentally observed gradual emergence of chromatin architecture. Our findings suggest a kinetic framework of chromatin architecture formation during zebrafish embryogenesis.
Transcriptional reprogramming by mutated IRF4 in lymphoma
Nikolai Schleussner, Pierre Cauchy, Vedran Franke, Maciej Giefing, Oriol Fornes, Naveen Vankadari, Salam A. Assi, Mariantonia Costanza, Marc A. Weniger, Altuna Akalin, Ioannis Anagnostopoulos, Thomas Bukur, Marco G. Casarotto, Frederik Damm, Oliver Daumke, Benjamin Edginton-White, J. Christof M. Gebhardt, Michael Grau, Stephan Grunwald, Martin-Leo Hansmann, Sylvia Hartmann, Lionel Huber, Eva Kärgel, Simone Lusatis, Daniel Noerenberg, Nadine Obier, Ulrich Pannicke, Anja Fischer, Anja Reisser, Andreas Rosenwald, Klaus Schwarz, Srinivasan Sundararaj, Andre Weilemann, Wiebke Winkler, Wendan Xu, Georg Lenz, Klaus Rajewsky, Wyeth W. Wasserman, Peter N. Cockerill, Claus Scheidereit, Reiner Siebert, Ralf Küppers, Rudolf Grosschedl, Martin Janz, Constanze Bonifer & Stephan Mathas
Disease-causing mutations in genes encoding transcription factors (TFs) can affect TF interactions with their cognate DNA-binding motifs. Whether and how TF mutations impact upon the binding to TF composite elements (CE) and the interaction with other TFs is unclear. Here, we report a distinct mechanism of TF alteration in human lymphomas with perturbed B cell identity, in particular classic Hodgkin lymphoma. It is caused by a recurrent somatic missense mutation c.295 T > C (p.Cys99Arg; p.C99R) targeting the center of the DNA-binding domain of Interferon Regulatory Factor 4 (IRF4), a key TF in immune cells. IRF4-C99R fundamentally alters IRF4 DNA-binding, with loss-of-binding to canonical IRF motifs and neomorphic gain-of-binding to canonical and non-canonical IRF CEs. IRF4-C99R thoroughly modifies IRF4 function by blocking IRF4-dependent plasma cell induction, and up-regulates disease-specific genes in a non-canonical Activator Protein-1 (AP-1)-IRF-CE (AICE)-dependent manner. Our data explain how a single mutation causes a complex switch of TF specificity and gene regulation and open the perspective to specifically block the neomorphic DNA-binding activities of a mutant TF.
https://doi.org/10.1038/s41467-023-41954-8
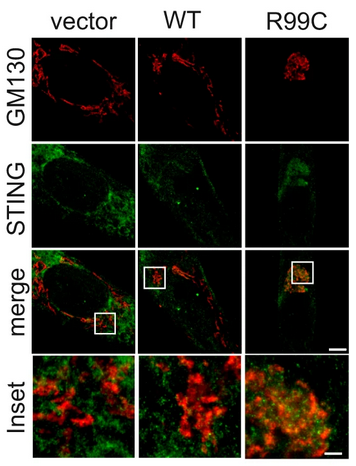
ARF1 prevents aberrant type I interferon induction by regulating STING activation and recycling
Maximilian Hirschenberger, Alice Lepelley, Ulrich Rupp, Susanne Klute, Victoria Hunszinger, Lennart Koepke, Veronika Merold, Blaise Didry-Barca, Fanny Wondany, Tim Bergner, Tatiana Moreau, Mathieu P. Rodero, Reinhild Rösler, Sebastian Wiese, Stefano Volpi, Marco Gattorno, Riccardo Papa, Sally-Ann Lynch, Marte G. Haug, Gunnar Houge, Kristen M. Wigby, Jessica Sprague, Jerica Lenberg, Clarissa Read, Paul Walther, Jens Michaelis, Frank Kirchhoff, Carina C. de Oliveira Mann, Yanick J. Crow & Konstantin M. J. Sparrer
Type I interferon (IFN) signalling is tightly controlled. Upon recognition of DNA by cyclic GMP-AMP synthase (cGAS), stimulator of interferon genes (STING) translocates along the endoplasmic reticulum (ER)-Golgi axis to induce IFN signalling. Termination is achieved through autophagic degradation or recycling of STING by retrograde Golgi-to-ER transport. Here, we identify the GTPase ADP-ribosylation factor 1 (ARF1) as a crucial negative regulator of cGAS-STING signalling. Heterozygous ARF1 missense mutations cause a previously unrecognized type I interferonopathy associated with enhanced IFN-stimulated gene expression. Disease-associated, GTPase-defective ARF1 increases cGAS-STING dependent type I IFN signalling in cell lines and primary patient cells. Mechanistically, mutated ARF1 perturbs mitochondrial morphology, causing cGAS activation by aberrant mitochondrial DNA release, and leads to accumulation of active STING at the Golgi/ERGIC due to defective retrograde transport. Our data show an unexpected dual role of ARF1 in maintaining cGAS-STING homeostasis, through promotion of mitochondrial integrity and STING recycling.
https://doi.org/10.1038/s41467-023-42150-4
Shank2 identifies a subset of glycinergic neurons involved in altered nociception in an autism model
Florian Olde Heuvel, Najwa Ouali Alami, Oumayma Aousji, Esther Pogatzki-Zahn, Peter K. Zahn, Hanna Wilhelm, Dhruva Deshpande, Elmira Khatamsaz, Alberto Catanese,
Sarah Woelfle, Michael Schön, Sanjay Jain, Stefanie Grabrucker, Albert C. Ludolph, Chiara Verpelli, Jens Michaelis, Tobias M. Boeckers and Francesco Roselli
Background Autism Spectrum Disorders (ASD) patients experience disturbed nociception in the form of either hyposensitivity to pain or allodynia. A substantial amount of processing of somatosensory and nociceptive stimulus takes place in the dorsal spinal cord. However, many of these circuits are not very well understood in the context of nociceptive processing in ASD.
Methods We have used a Shank2−/− mouse model, which displays a set of phenotypes reminiscent of ASD, and performed behavioural and microscopic analysis to investigate the role of dorsal horn circuitry in nociceptive processing of ASD.
Results We determined that Shank2−/− mice display increased sensitivity to formalin pain and thermal preference, but a sensory specific mechanical allodynia. We demonstrate that high levels of Shank2 expression identifies a subpopulation of neurons in murine and human dorsal spinal cord, composed mainly by glycinergic interneurons and that loss of Shank2 causes the decrease in NMDAR in excitatory synapses on these inhibitory interneurons. In fact, in the subacute phase of the formalin test, glycinergic interneurons are strongly activated in wild type (WT) mice but not in Shank2−/− mice. Consequently, nociception projection neurons in laminae I are activated in larger numbers in Shank2−/− mice.
Limitations Our investigation is limited to male mice, in agreement with the higher representation of ASD in males; therefore, caution should be applied to extrapolate the findings to females. Furthermore, ASD is characterized by extensive genetic diversity and therefore the findings related to Shank2 mutant mice may not necessarily apply to patients with different gene mutations. Since nociceptive phenotypes in ASD range between hyper- and hypo-sensitivity, diverse mutations may affect the circuit in opposite ways.
https://doi.org/10.1186/s13229-023-00552-7
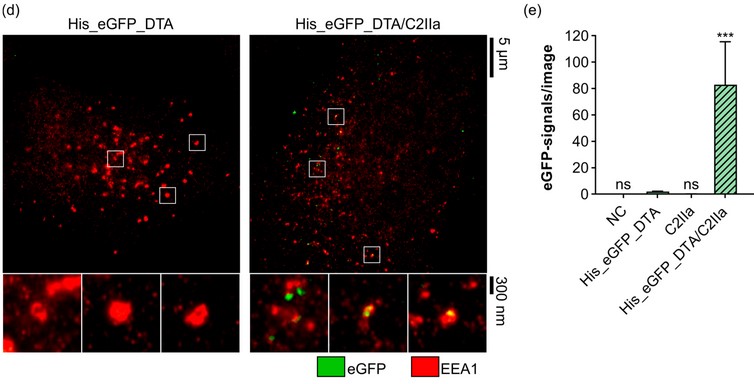
The Clostridium botulinum C2 Toxin Subunit C2IIa Delivers Enzymes with Positively Charged N-Termini into the Cytosol of Target Cells
Sebastian Heber, Joscha Borho, Nicole Stadler, Fanny Wondany, Irina König, Jens Michaelis, Panagiotis Papatheodorou, Holger Barth and Maximilian Fellermann
The binary Clostridium (C.) botulinum C2 toxin consists of two non-linked proteins. The proteolytically activated binding/transport subunit C2IIa forms barrel-shaped homoheptamers, which bind to cell surface receptors, mediate endocytosis, and translocate the enzyme subunit C2I into the cytosol of target cells. Here, we investigate whether C2IIa can be harnessed as a transporter for proteins/enzymes fused to polycationic tags, as earlier demonstrated for the related anthrax toxin transport subunit PA63. To test C2IIa-mediated transport in cultured cells, reporter enzymes are generated by fusing different polycationic tags to the N- or C-terminus of other bacterial toxins’ catalytic A subunits. C2IIa as well as PA63 deliver N-terminally polyhistidine-tagged proteins more efficiently compared to C-terminally tagged ones. However, in contrast to PA63, C2IIa does not efficiently deliver polylysine-tagged proteins into the cytosol of target cells. Moreover, untagged enzymes with a native cationic N-terminus are efficiently transported by both C2IIa and PA63. In conclusion, the C2IIa-transporter serves as a transport system for enzymes that harbor positively charged amino acids at their N-terminus. The charge distribution at the N-terminus of cargo proteins and their ability to unfold in the endosome and subsequently refold in the cytosol determine transport feasibility and efficiency.
https://doi.org/10.3390/toxins15060390
Reliability and accuracy of single-molecule FRET studies for characterization of structural dynamics and distances in proteins
Ganesh Agam, Christian Gebhardt, Milana Popara,..., Jens Michaelis,..., Anna Sefer,..., et al.
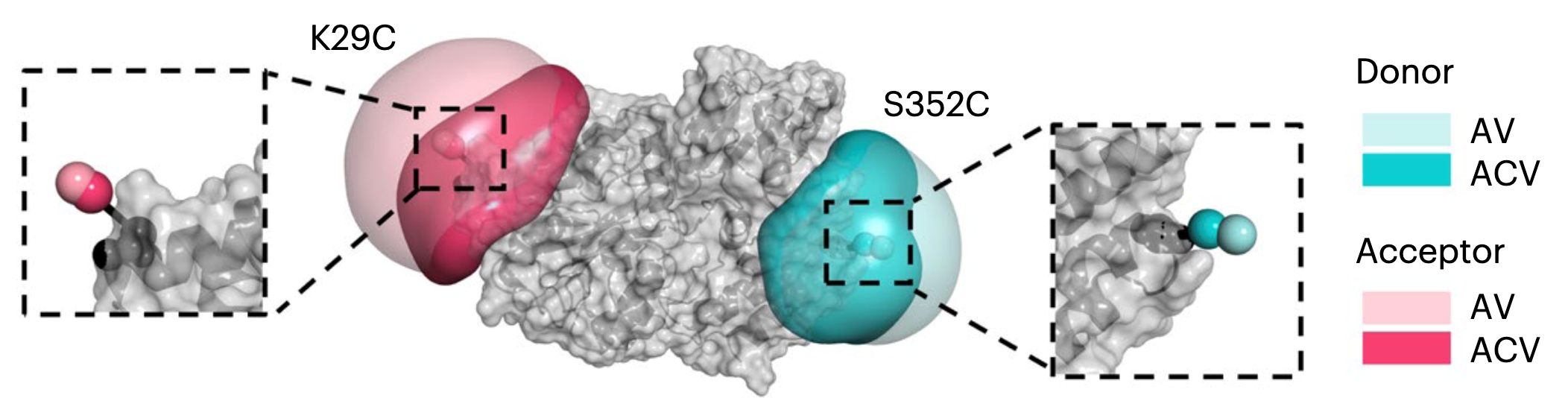
Single-molecule Förster-resonance energy transfer (smFRET) experiments allow the study of biomolecular structure and dynamics in vitro and in vivo. We performed an international blind study involving 19 laboratories to assess the uncertainty of FRET experiments for proteins with respect to the measured FRET efficiency histograms, determination of distances, and the detection and quantification of structural dynamics. Using two protein systems with distinct conformational changes and dynamics, we obtained an uncertainty of the FRET efficiency ≤0.06, corresponding to an interdye distance precision of ≤2 Å and accuracy of ≤5 Å. We further discuss the limits for detecting fluctuations in this distance range and how to identify dye perturbations. Our work demonstrates the ability of smFRET experiments to simultaneously measure distances and avoid the averaging of conformational dynamics for realistic protein systems, highlighting its importance in the expanding toolbox of integrative structural biology. https://www.nature.com/articles/s41592-023-01807-0
A multimorphic mutation in IRF4 causes human autosomal dominant combined immunodeficiency
Christof M. Gebhardt and IRF4 International Consortium authors and contributions
Interferon regulatory factor 4 (IRF4) is a transcription factor (TF) and key regulator of immune cell development and function. We report a recurrent heterozygous mutation in IRF4, p.T95R, causing an autosomal dominant combined immunodeficiency (CID) in seven patients from six unrelated families. The patients exhibited profound susceptibility to opportunistic infections, notably Pneumocystis jirovecii, and presented with agammaglobulinemia. Patients’ B cells showed impaired maturation, decreased immunoglobulin isotype switching, and defective plasma cell differentiation, whereas their T cells contained reduced TH17 and TFH populations and exhibited decreased cytokine production. A knock-in mouse model of heterozygous T95R showed a severe defect in antibody production both at the steady state and after immunization with different types of antigens, consistent with the CID observed in these patients. The IRF4T95R variant maps to the TF’s DNA binding domain, alters its canonical DNA binding specificities, and results in a simultaneous multimorphic combination of loss, gain, and new functions for IRF4. IRF4T95R behaved as a gain-of-function hypermorph by binding to DNA with higher affinity than IRF4WT. Despite this increased affinity for DNA, the transcriptional activity on IRF4 canonical genes was reduced, showcasing a hypomorphic activity of IRF4T95R. Simultaneously, IRF4T95R functions as a neomorph by binding to noncanonical DNA sites to alter the gene expression profile, including the transcription of genes exclusively induced by IRF4T95R but not by IRF4WT. This previously undescribed multimorphic IRF4 pathophysiology disrupts normal lymphocyte biology, causing human disease.
https://doi.org/10.1126/sciimmunol.ade7953