Publikationen 2021
Transcription Factor RBPJL Is Able to Repress Notch Target Gene Expression but Is Non-Responsive to Notch Activation
Philipp Hoffmeister, Aleksandra Turkiewicz, N. N. Duyen Huynh, Andreas Große-Berkenbusch, Uwe Knippschild, J. Christof M. Gebhardt, Bernd Baumann, Tilman Borggrefe, Franz Oswald
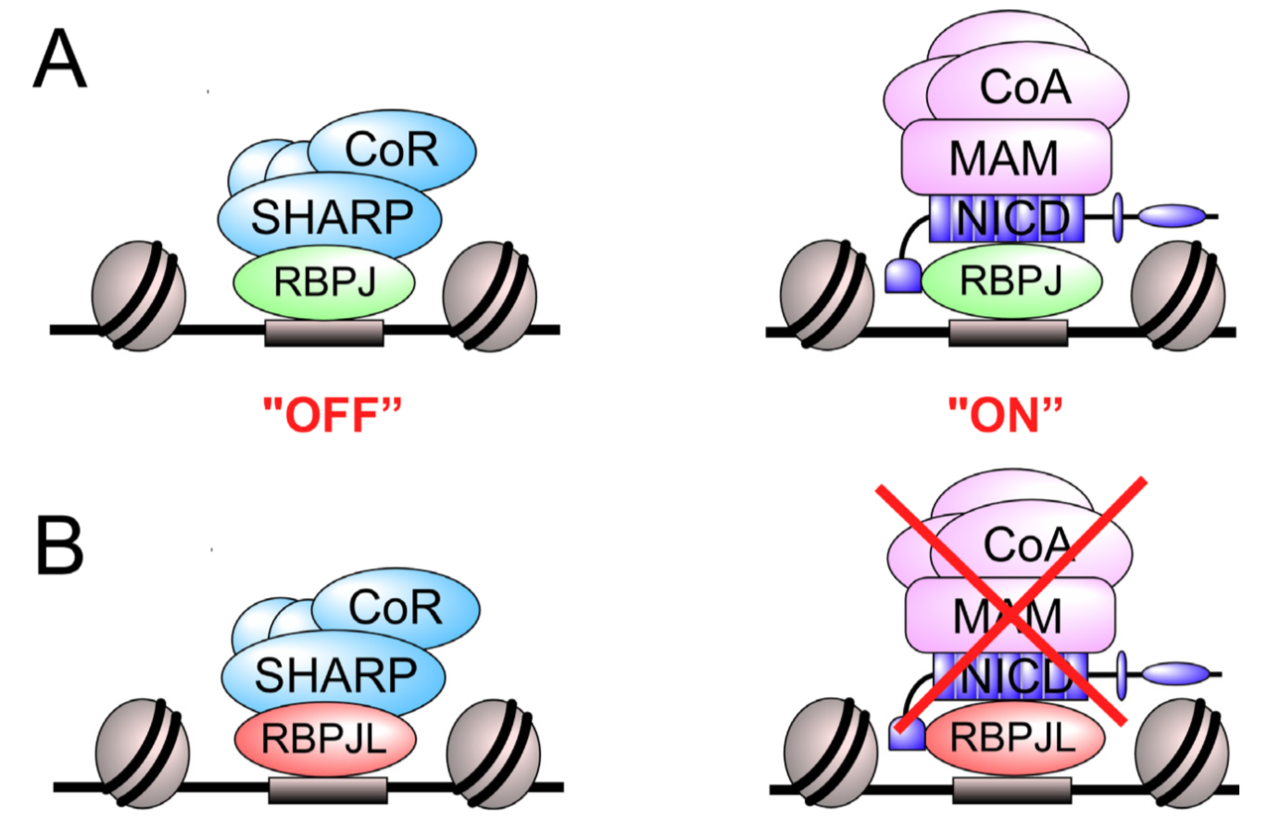
The Notch signaling pathway is an evolutionary conserved signal transduction cascade present in almost all tissues and is required for embryonic and postnatal development, as well as for stem cell maintenance, but it is also implicated in tumorigenesis including pancreatic cancer and leukemia. The transcription factor RBPJ forms a coactivator complex in the presence of a Notch signal, whereas it represses Notch target genes in the absence of a Notch stimulus. In the pancreas, a specific paralog of RBPJ, called RBPJL, is expressed and found as part of the heterotrimeric PTF1-complex. However, the function of RBPJL in Notch signaling remains elusive. Using molecular modeling, biochemical and functional assays, as well as single-molecule time-lapse imaging, we show that RBPJL and RBPJ, despite limited sequence homology, possess a high degree of structural similarity. RBPJL is specifically expressed in the exocrine pancreas, whereas it is mostly undetectable in pancreatic tumour cell lines. Importantly, RBPJL is not able to interact with Notch−1 to −4 and it does not support Notch-mediated transactivation. However, RBPJL can bind to canonical RBPJ DNA elements and shows migration dynamics comparable to that of RBPJ in the nuclei of living cells. Importantly, RBPJL is able to interact with SHARP/SPEN, the central corepressor of the Notch pathway. In line with this, RBPJL is able to fully reconstitute transcriptional repression at Notch target genes in cells lacking RBPJ. Together, RBPJL can act as an antagonist of RBPJ, which renders cells unresponsive to the activation of Notch.
https://www.mdpi.com/2072-6694/13/19/5027
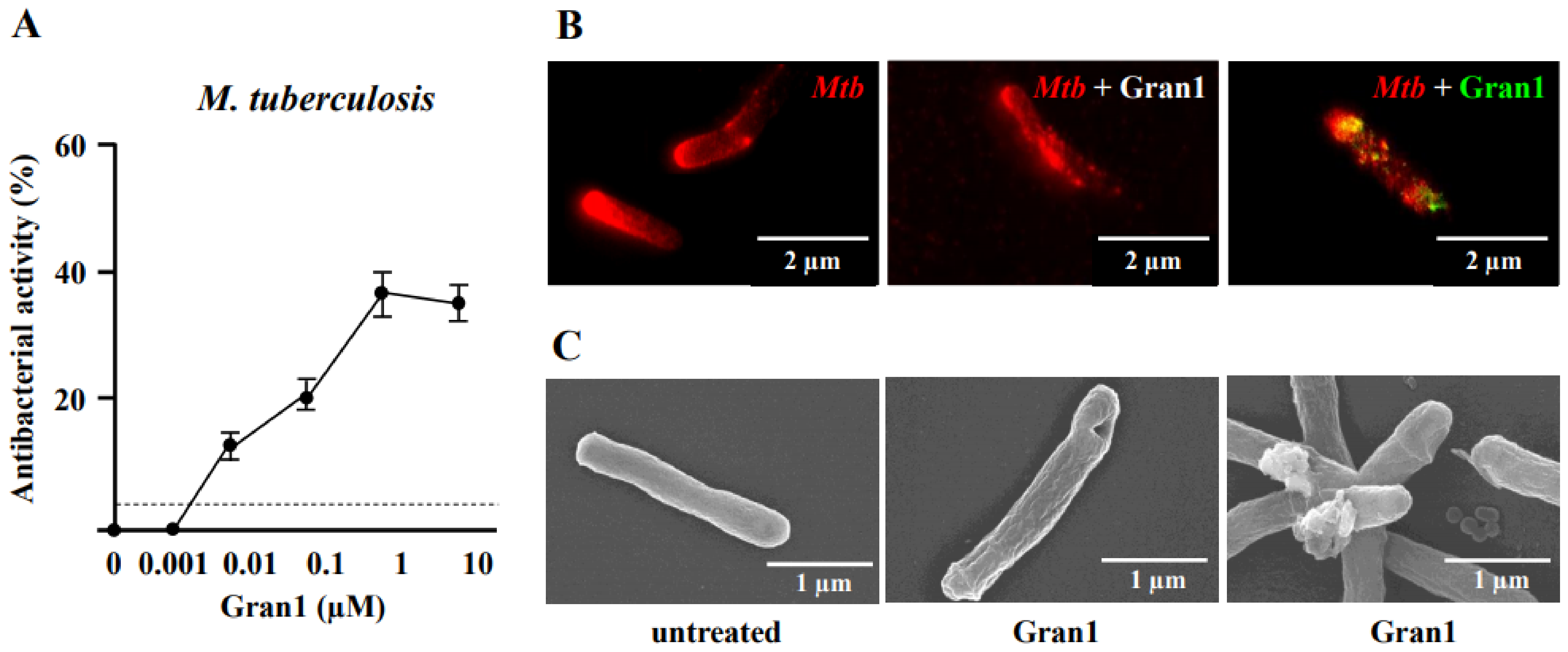
A Granulysin-Derived Peptide with Potent Activity against Intracellular Mycobacterium tuberculosis
Reiner Noschka, Fanny Wondany, Gönül Kizilsavas, Tanja Weil, Gilbert Weidinger, Paul Walther, Jens Michaelis, Steffen Stenger
Granulysin is an antimicrobial peptide (AMP) expressed by human T-lymphocytes and natural killer cells. Despite a remarkably broad antimicrobial spectrum, its implementation into clinical practice has been hampered by its large size and off-target effects. To circumvent these limitations, we synthesized a 29 amino acid fragment within the putative cytolytic site of Granulysin (termed “Gran1”). We evaluated the antimicrobial activity of Gran1 against the major human pathogen Mycobacterium tuberculosis (Mtb) and a panel of clinically relevant non-tuberculous mycobacteria which are notoriously difficult to treat. Gran1 efficiently inhibited the mycobacterial proliferation in the low micro molar range. Super-resolution fluorescence microscopy and scanning electron microscopy indicated that Gran1 interacts with the surface of Mtb, causing lethal distortions of the cell wall. Importantly, Gran1 showed no off-target effects (cytokine release, chemotaxis, cell death) in primary human cells or zebrafish embryos (cytotoxicity, developmental toxicity, neurotoxicity, cardiotoxicity). Gran1 was selectively internalized by macrophages, the major host cell of Mtb, and restricted the proliferation of the pathogen. Our results demonstrate that the hypothesis-driven design of AMPs is a powerful approach for the identification of small bioactive compounds with specific antimicrobial activity. Gran1 is a promising component for the design of AMP-containing nanoparticles with selective activity and favorable pharmacokinetics to be pushed forward into experimental in vivo models of infectious diseases, most notably tuberculosis.
https://www.mdpi.com/1422-0067/22/16/8392
A guide to changing paradigms of glucocorticoid receptor function—a model system for genome regulation and physiology
Sabine Vettorazzi, Denis Nalbantoglu, J. Christof M. Gebhardt, Jan Tuckermann
The glucocorticoid receptor (GR) is a bona fide ligand-regulated transcription factor. Cloned in the 80s, the GR has become one of the best-studied and clinically most relevant members of the nuclear receptor superfamily. Cooperative activity of GR with other transcription factors and a plethora of coregulators contribute to the tissue- and context-specific response toward the endogenous and pharmacological glucocorticoids (GCs). Furthermore, nontranscriptional activities in the cytoplasm are emerging as an additional function of GR. Over the past 40 years, the concepts of GR mechanisms of action had been constantly changing. Different methodologies in the pregenomic and genomic era of molecular biological research and recent cutting-edge technology in single-cell and single-molecule analysis are steadily evolving the views, how the GR in particular and transcriptional regulation in general act in physiological and pathological processes. In addition to the development of technologies for GR analysis, the use of model organisms provides insights how the GR in vivo executes GC action in tissue homeostasis, inflammation, and energy metabolism. The model organisms, namely the mouse, but also rats, zebrafish, and recently fruit flies carrying mutations of the GR became a major driving force to analyze the molecular function of GR in disease models. This guide provides an overview of the exciting research and paradigm shifts in the GR field from past to present with a focus on GR transcription factor networks, GR DNA-binding and single-cell analysis, and model systems.
https://doi.org/10.1111/febs.16100
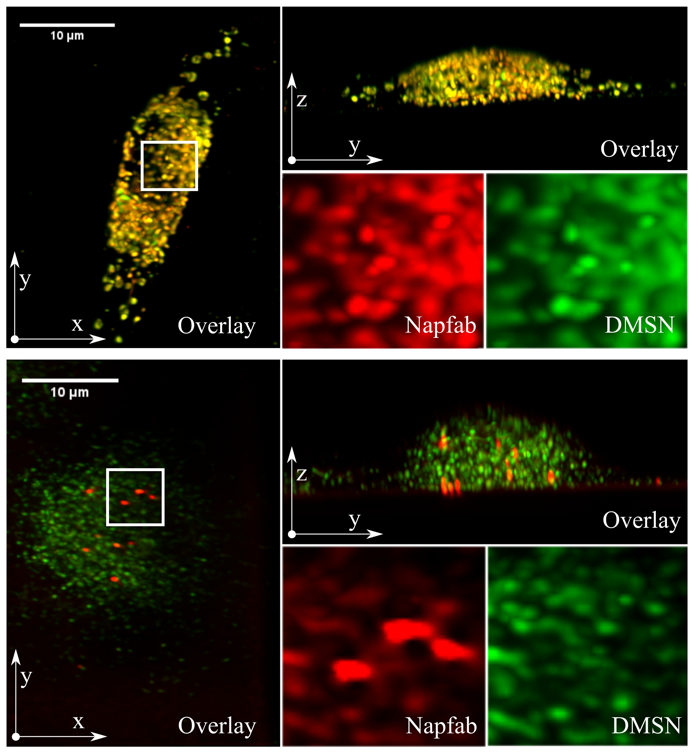
Delivery by Dendritic Mesoporous Silica Nanoparticles Enhances the Antimicrobial Activity of a Napsin-Derived Peptide Against Intracellular Mycobacterium tuberculosis
Bastian Beitzinger, Fabian Gerbl, Thomas Vomhof, Roman Schmid, Reiner Noschka, Armando Rodriguez, Sebastian Wiese, Gilbert Weidinger, Ludger Ständker, Paul Walther, Jens Michaelis, Mika Lindén, Steffen Stenger
Tuberculosis remains a serious global health problem causing 1.3 million deaths annually. The causative pathogen Mycobacterium tuberculosis (Mtb) has developed several mechanisms to evade the immune system and resistances to many conventional antibiotics, so that alternative treatment strategies are urgently needed. By isolation from bronchoalveolar lavage and peptide optimization, a new antimicrobial peptide named NapFab is discovered. While showing robust activity against extracellular Mtb, the activity of NapFab against intracellular bacteria is limited due to low intracellular availability. By loading NapFab onto dendritic mesoporous silica nanoparticles (DMSN) as a carrier system, cellular uptake, and consequently antimycobacterial activity against intracellular Mtb is significantly enhanced. Furthermore, using lattice light-sheet fluorescence microscopy, it can be shown that the peptide is gradually released from the DMSN inside living macrophages over time. By electron microscopy and tomography, it is demonstrated that peptide loaded DMSN are stored in vesicular structures in proximity to mycobacterial phagosomes inside the cells, but the nanoparticles are typically not in direct contact with the bacteria. Based on the combination of functional and live-cell imaging analyses, it is hypothesized that after being released from the DMSN NapFab is able to enter the bacterial phagosome and gain access to the bacilli.
https://doi.org/10.1002/adhm.202100453
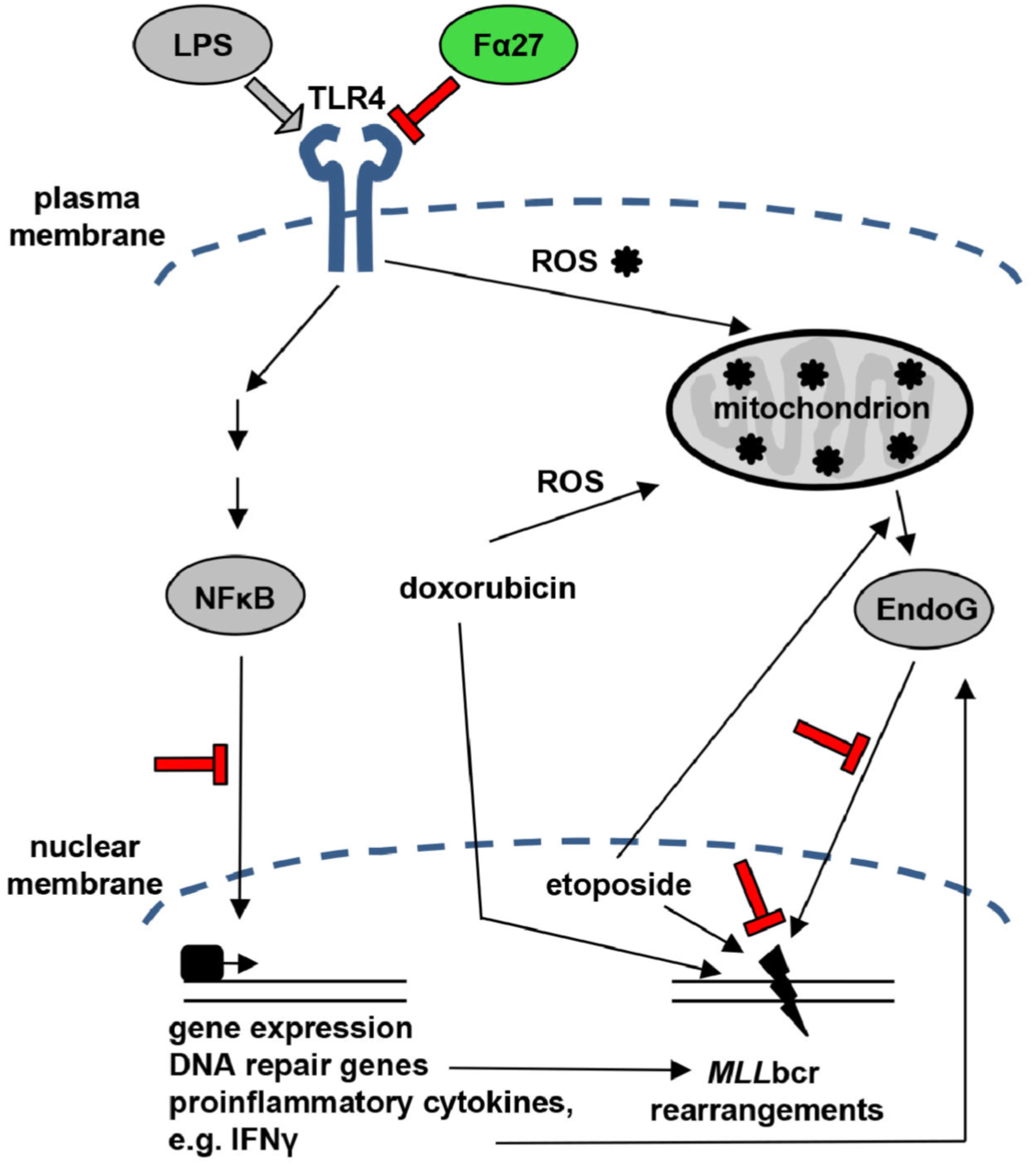
A Fibrinogen Alpha Fragment Mitigates Chemotherapy-Induced MLL Rearrangements
Eberle Julia, Wiehe Rahel Stefanie, Gole Boris, Mattis Liska Jule, Palmer Anja, Ständker Ludger, Forssmann Wolf-Georg, Münch Jan, Gebhardt J. Christof M., Wiesmüller Lisa
Rearrangements in the Mixed Lineage Leukemia breakpoint cluster region (MLLbcr) are frequently involved in therapy-induced leukemia, a severe side effect of anti-cancer therapies. Previous work unraveled Endonuclease G as the critical nuclease causing initial breakage in the MLLbcr in response to different types of chemotherapeutic treatment. To identify peptides protecting against therapy-induced leukemia, we screened a hemofiltrate-derived peptide library by use of an enhanced green fluorescent protein (EGFP)-based chromosomal reporter of MLLbcr rearrangements. Chromatographic purification of one active fraction and subsequent mass spectrometry allowed to isolate a C-terminal 27-mer of fibrinogen α encompassing amino acids 603 to 629. The chemically synthesized peptide, termed Fα27, inhibited MLLbcr rearrangements in immortalized hematopoietic cells following treatment with the cytostatics etoposide or doxorubicin. We also provide evidence for protection of primary human hematopoietic stem and progenitor cells from therapy-induced MLLbcr breakage. Of note, fibrinogen has been described to activate toll-like receptor 4 (TLR4). Dissecting the Fα27 mode-of action revealed association of the peptide with TLR4 in an antagonistic fashion affecting downstream NFκB signaling and pro-inflammatory cytokine production. In conclusion, we identified a hemofiltrate-derived peptide inhibitor of the genome destabilizing events causing secondary leukemia in patients undergoing chemotherapy.
https://www.frontiersin.org/article/10.3389/fonc.2021.689063
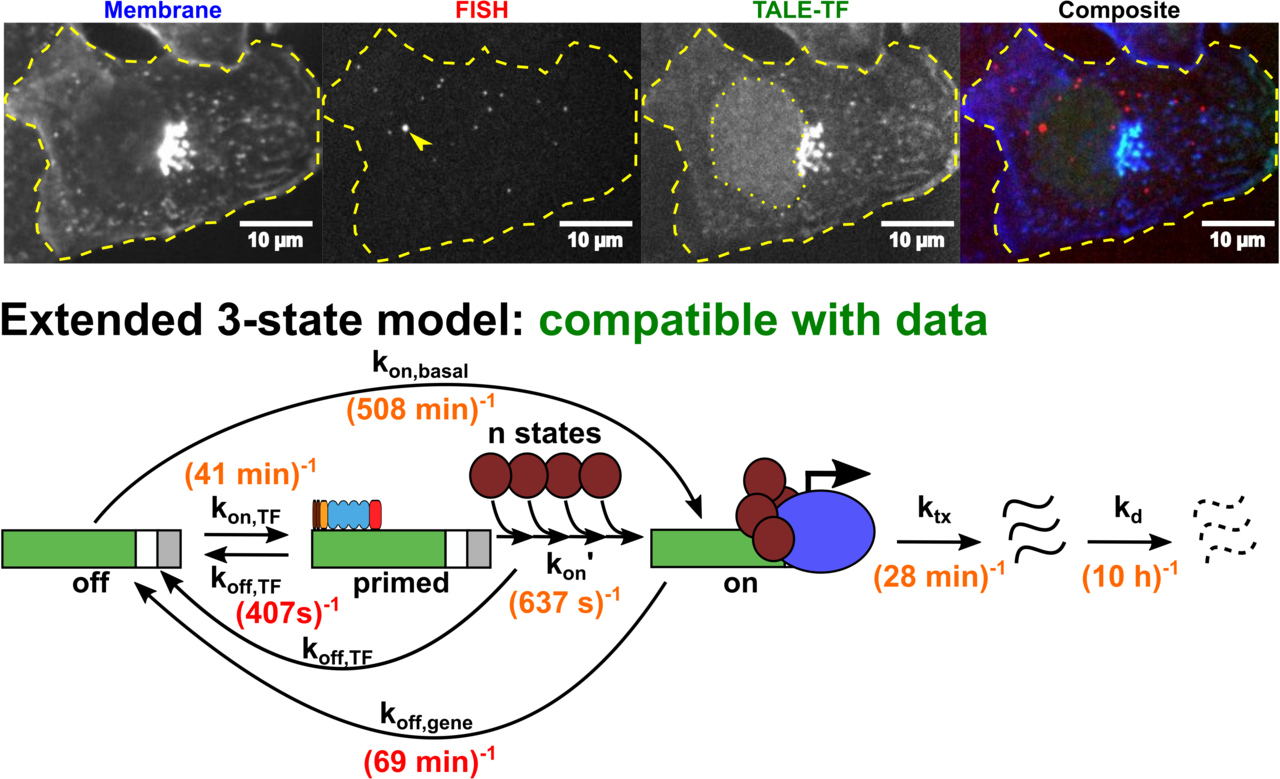
Altering transcription factor binding reveals comprehensive transcriptional kinetics of a basic gene
Transcription is a vital process activated by transcription factor (TF) binding. The active gene releases a burst of transcripts before turning inactive again. While the basic course of transcription is well understood, it is unclear how binding of a TF affects the frequency, duration and size of a transcriptional burst. We systematically varied the residence time and concentration of a synthetic TF and characterized the transcription of a synthetic reporter gene by combining single molecule imaging, single molecule RNA-FISH, live transcript visualisation and analysis with a novel algorithm, Burst Inference from mRNA Distributions (BIRD). For this well-defined system, we found that TF binding solely affected burst frequency and variations in TF residence time had a stronger influence than variations in concentration. This enabled us to device a model of gene transcription, in which TF binding triggers multiple successive steps before the gene transits to the active state and actual mRNA synthesis is decoupled from TF presence. We quantified all transition times of the TF and the gene, including the TF search time and the delay between TF binding and the onset of transcription. Our quantitative measurements and analysis revealed detailed kinetic insight, which may serve as basis for a bottom-up understanding of gene regulation.
https://doi.org/10.1093/nar/gkab443
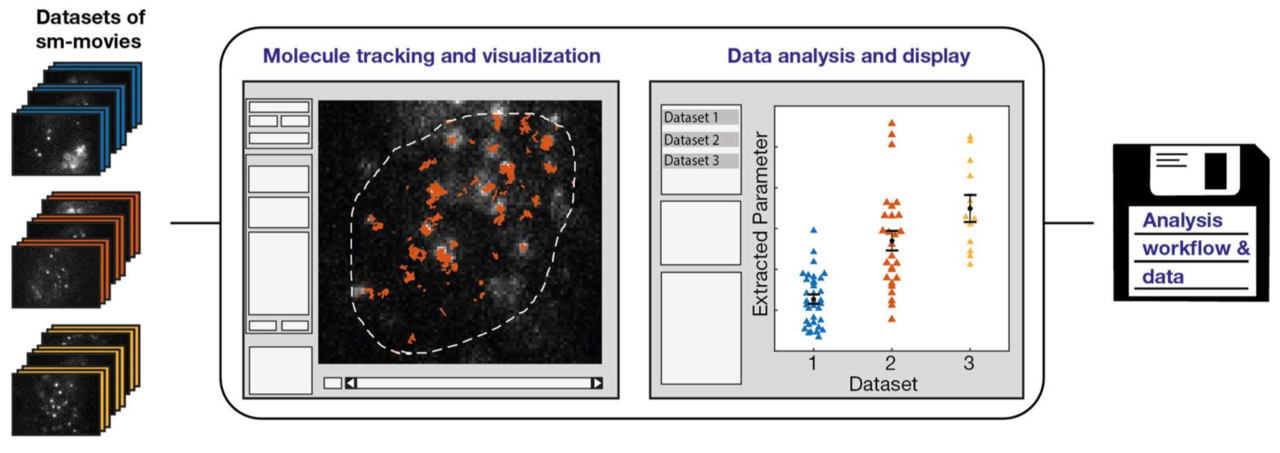
Single molecule tracking and analysis framework including theory-predicted parameter settings
Rubina Davtyan,
Imaging, tracking and analyzing individual biomolecules in living systems is a powerful technology to obtain quantitative kinetic and spatial information such as reaction rates, diffusion coefficients and localization maps. Common tracking tools often operate on single movies and require additional manual steps to analyze whole data sets or to compare different experimental conditions. We report a fast and comprehensive single molecule tracking and analysis framework (TrackIt) to simultaneously process several multi-movie data sets. A user-friendly GUI offers convenient tracking visualization, multiple state-of-the-art analysis procedures, display of results, and data im- and export at different levels to utilize external software tools. We applied our framework to quantify dissociation rates of a transcription factor in the nucleus and found that tracking errors, similar to fluorophore photobleaching, have to be considered for reliable analysis. Accordingly, we developed an algorithm, which accounts for both tracking losses and suggests optimized tracking parameters when evaluating reaction rates. Our versatile and extensible framework facilitates quantitative analysis of single molecule experiments at different experimental conditions.
https://doi.org/10.1038/s41598-021-88802-7
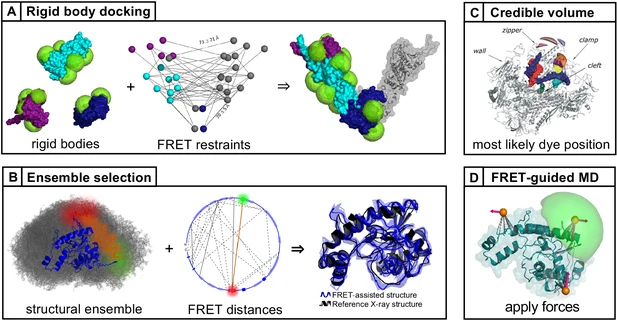
FRET-based dynamic structural biology: Challenges, perspectives and an appeal for open-science practices
Eitan Lerner, Anders Barth, Jelle Hendrix, Benjamin Ambrose, Victoria Birkedal, Scott C Blanchard, Richard Börner, Hoi Sung Chung, Thorben Cordes, Timothy D Craggs, Ashok A Deniz, Jiajia Diao, Jingyi Fei, Ruben L Gonzalez, Irina V Gopich, Taekjip Ha, Christian A Hanke, Gilad Haran, Nikos S Hatzakis, Sungchul Hohng, Seok-Cheol Hong, Thorsten Hugel, Antonino Ingargiola, Chirlmin Joo, Achillefs N Kapanidis, Harold D Kim, Ted Laurence, Nam Ki Lee, Tae-Hee Lee, Edward A Lemke, Emmanuel Margeat, Jens Michaelis, Xavier Michalet, Sua Myong, Daniel Nettels, Thomas-Otavio Peulen, Evelyn Ploetz, Yair Razvag, Nicole C Robb, Benjamin Schuler, Hamid Soleimaninejad, Chun Tang, Reza Vafabakhsh, Don C Lamb, Claus AM Seidel, Shimon Weiss
Single-molecule FRET (smFRET) has become a mainstream technique for studying biomolecular structural dynamics. The rapid and wide adoption of smFRET experiments by an ever-increasing number of groups has generated significant progress in sample preparation, measurement procedures, data analysis, algorithms and documentation. Several labs that employ smFRET approaches have joined forces to inform the smFRET community about streamlining how to perform experiments and analyze results for obtaining quantitative information on biomolecular structure and dynamics. The recent efforts include blind tests to assess the accuracy and the precision of smFRET experiments among different labs using various procedures. These multi-lab studies have led to the development of smFRET procedures and documentation, which are important when submitting entries into the archiving system for integrative structure models, PDB-Dev. This position paper describes the current ‘state of the art’ from different perspectives, points to unresolved methodological issues for quantitative structural studies, provides a set of ‘soft recommendations’ about which an emerging consensus exists, and lists openly available resources for newcomers and seasoned practitioners. To make further progress, we strongly encourage ‘open science’ practices.
https://elifesciences.org/articles/60416
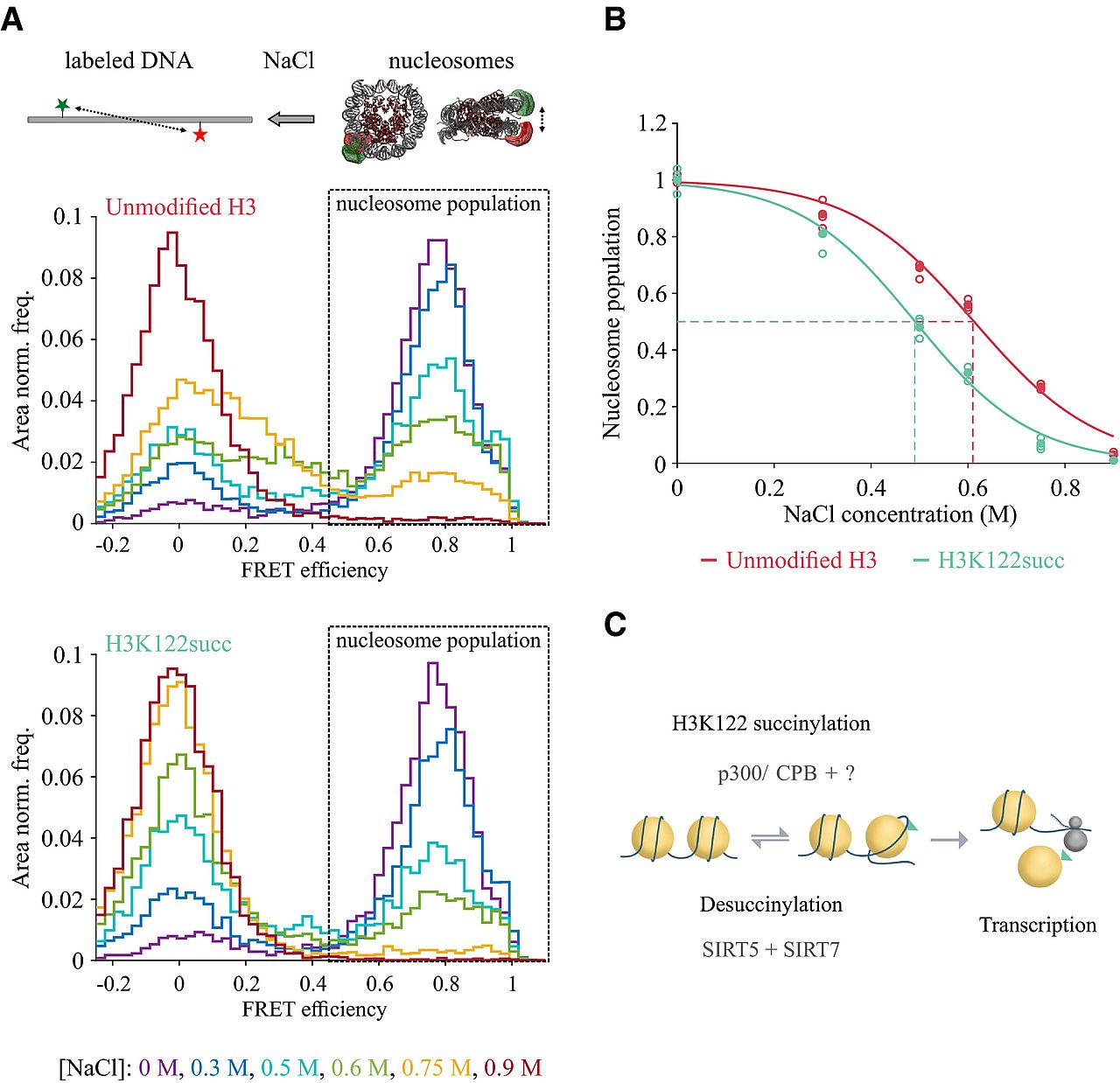
Succinylation of H3K122 destabilizes nucleosomes and enhances transcription
Lara Zorro Shahidian, Mariska Haas, Stephanie Le Gras, Sandra Nitsch, André Mourão, Arie Geerlof, Raphael Margueron, Jens Michaelis, Sylvain Daujat, Robert Schneider
Histone post‐translational modifications (PTMs) are key players in chromatin regulation. The identification of novel histone acylations raises important questions regarding their role in transcription. In this study, we characterize the role of an acylation on the lateral surface of the histone octamer, H3K122 succinylation (H3K122succ), in chromatin function and transcription. Using chromatin succinylated at H3K122 in in vitro transcription assays, we show that the presence of H3K122succ is sufficient to stimulate transcription. In line with this, we found in our ChIP assays H3K122succ enriched on promoters of active genes and H3K122succ enrichment scaling with gene expression levels. Furthermore, we show that the co‐activators p300/CBP can succinylate H3K122 and identify sirtuin 5 (SIRT5) as a new desuccinylase. By applying single molecule FRET assays, we demonstrate a direct effect of H3K122succ on nucleosome stability, indicating an important role for histone succinylation in modulating chromatin dynamics. Together, these data provide the first insights into the mechanisms underlying transcriptional regulation by H3K122succ.
https://www.embopress.org/doi/full/10.15252/embr.202051009
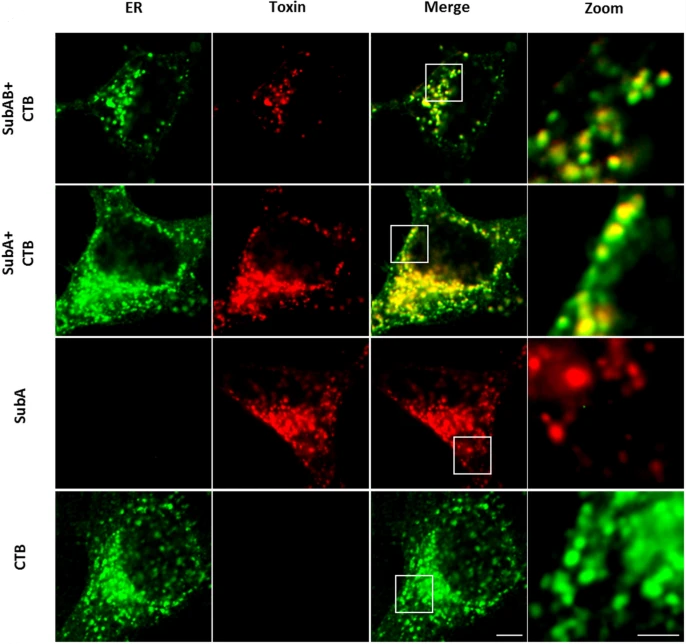
The enzyme subunit SubA of Shiga toxin-producing E. coli strains demonstrates comparable intracellular transport and cytotoxic activity as the holotoxin SubAB in HeLa and HCT116 cells in vitro
Katharina Sessler, Panagiotis Papatheodorou, Fanny Wondany, Maike Krause, Sabrina Noettger, Denise Bernhard, Jens Michaelis, Herbert Schmidt & Holger Barth
The subtilase cytotoxin (SubAB) is secreted by certain Shiga toxin-producing Escherichia coli (STEC) strains and is composed of the enzymatically active subunit SubA and the pentameric binding/transport subunit SubB. We previously demonstrated that SubA (10 µg/ml), in the absence of SubB, binds and intoxicates the human cervix cancer-derived epithelial cell line HeLa. However, the cellular and molecular mechanisms underlying the cytotoxic activity of SubA in the absence of SubB remained unclear. In the present study, the cytotoxic effects mediated by SubA alone were investigated in more detail in HeLa cells and the human colon cancer cell line HCT116. We found that in the absence of SubB, SubA (10 µg/ml) is internalized into the endoplasmic reticulum (ER), where it cleaves the chaperone GRP78, an already known substrate for SubA after its canonical uptake into cells via SubB. The autonomous cellular uptake of SubA and subsequent cleavage of GRP78 in cells is prevented by treatment of cells with 10 µM brefeldin A, which inhibits the transport of protein toxins into the ER. In addition, by analyzing the SubA mutant SubAΔC344, we identified the C-terminal SEEL motif as an ER-targeting signal. Conclusively, our results strongly suggest that SubA alone shares the same intracellular transport route and cytotoxic activity as the SubAB holotoxin.
https://link.springer.com/article/10.1007/s00204-020-02965-2