Analysis tools
Published projects:
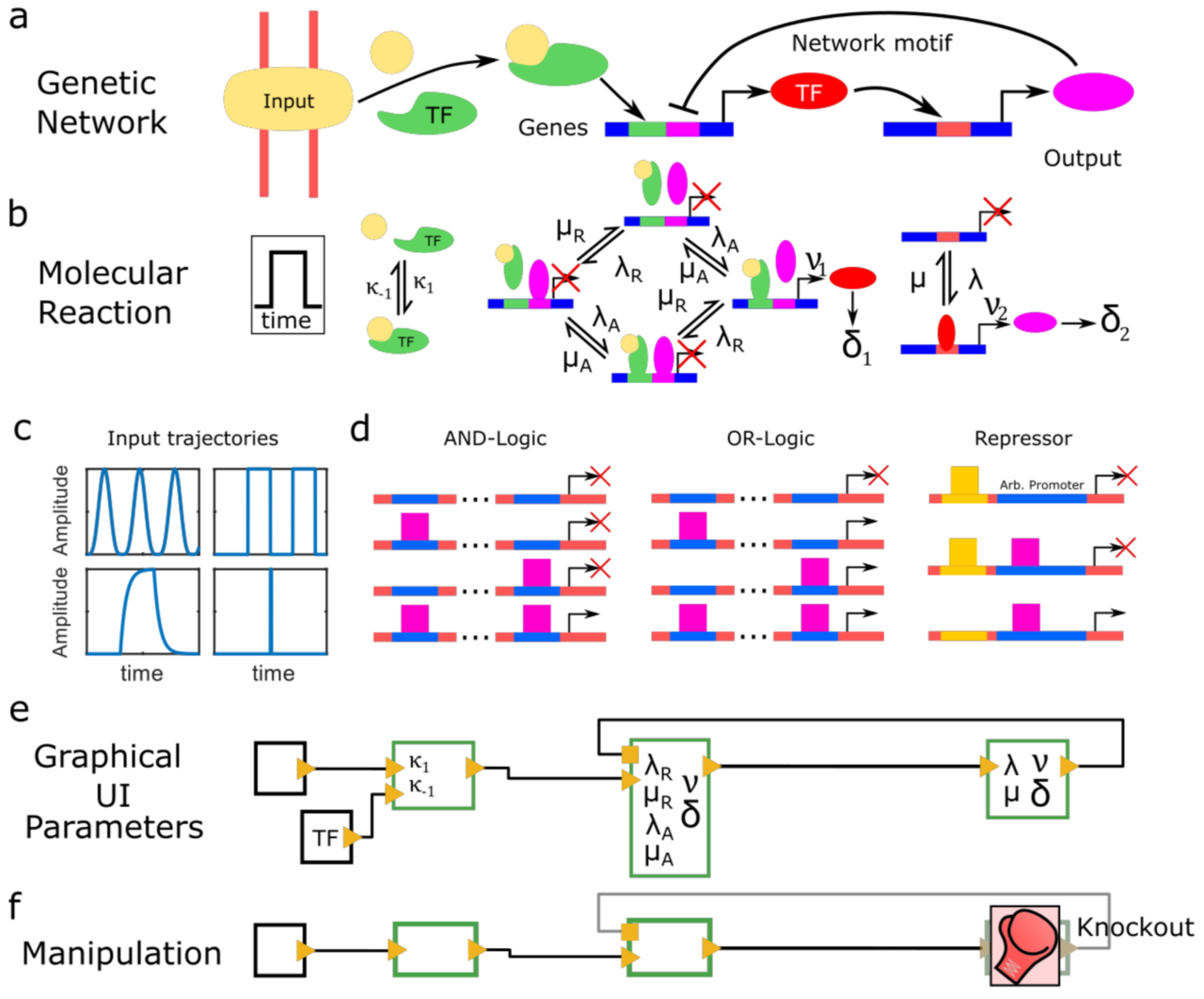
Periodic synchronization of isolated network elements facilitates simulating and inferring gene regulatory networks including stochastic molecular kinetics
The temporal progression of many fundamental processes in cells and organisms, including homeostasis, differentiation and development, are governed by gene regulatory networks (GRNs). GRNs balance fluctuations in the output of their genes, which trace back to the stochasticity of molecular interactions. Although highly desirable to understand life processes, predicting the temporal progression of gene products within a GRN is challenging when considering stochastic events such as transcription factor–DNA interactions or protein production and degradation.
We report a method to simulate and infer GRNs including genes and biochemical reactions at molecular detail. In our approach, we consider each network element to be isolated from other elements during small time intervals, after which we synchronize molecule numbers across all network elements. Thereby, the temporal behaviour of network elements is decoupled and can be treated by local stochastic or deterministic solutions. We demonstrate the working principle of this modular approach with a repressive gene cascade comprising four genes. By considering a deterministic time evolution within each time interval for all elements, our method approaches the solution of the system of deterministic differential equations associated with the GRN. By allowing genes to stochastically switch between on and off states or by considering stochastic production of gene outputs, we are able to include increasing levels of stochastic detail and approximate the solution of a Gillespie simulation. Thereby, CaiNet is able to reproduce noise-induced bi-stability and oscillations in dynamically complex GRNs. Notably, our modular approach further allows for a simple consideration of deterministic delays. We further infer relevant regulatory connections and steady-state parameters of a GRN of up to ten genes from steady-state measurements by identifying each gene of the network with a single perceptron in an artificial neuronal network and using a gradient decent method originally designed to train recurrent neural networks. To facilitate setting up GRNs and using our simulation and inference method, we provide a fast computer-aided interactive network simulation environment, CaiNet.
We developed a method to simulate GRNs at molecular detail and to infer the topology and steady-state parameters of GRNs. Our method and associated user-friendly framework CaiNet should prove helpful to analyze or predict the temporal progression of reaction networks or GRNs in cellular and organismic biology. CaiNet is freely available at https://gitlab.com/GebhardtLab/CaiNet.
Johannes Hettich, Christof M. Gebhardt
https://doi.org/10.1186/s12859-021-04541-6
Single molecule tracking and analysis framework including theory-predicted parameter settings
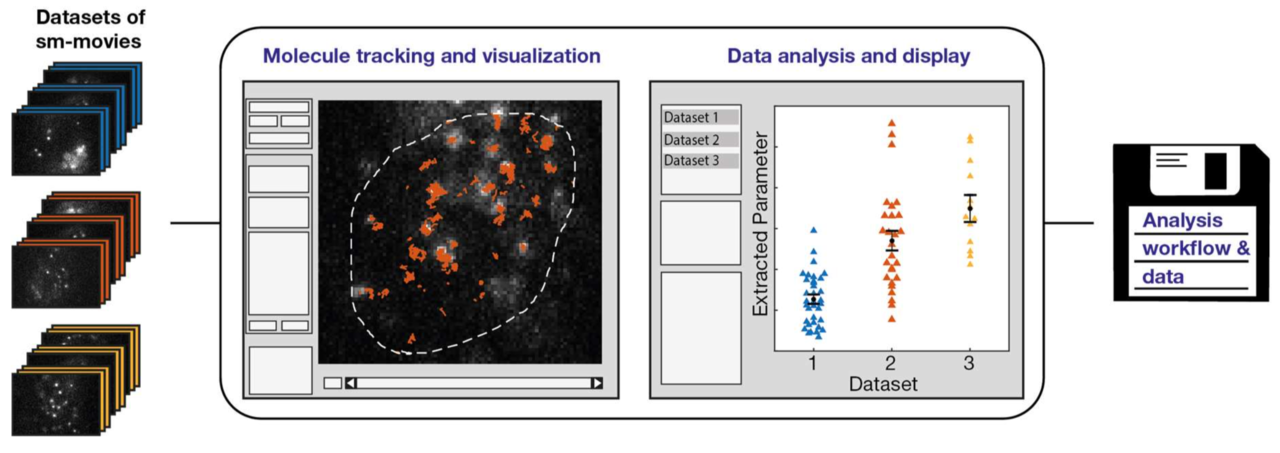
Imaging, tracking and analyzing individual biomolecules in living systems is a powerful technology to obtain quantitative kinetic and spatial information such as reaction rates, diffusion coefficients and localization maps. Common tracking tools often operate on single movies and require additional manual steps to analyze whole data sets or to compare different experimental conditions. We report a fast and comprehensive single molecule tracking and analysis framework (TrackIt) to simultaneously process several multi-movie data sets. A user-friendly GUI offers convenient tracking visualization, multiple state-of-the-art analysis procedures, display of results, and data im- and export at different levels to utilize external software tools. We applied our framework to quantify dissociation rates of a transcription factor in the nucleus and found that tracking errors, similar to fluorophore photobleaching, have to be considered for reliable analysis. Accordingly, we developed an algorithm, which accounts for both tracking losses and suggests optimized tracking parameters when evaluating reaction rates. Our versatile and extensible framework facilitates quantitative analysis of single molecule experiments at different experimental conditions.
Rubina Davtyan,
https://doi.org/10.1038/s41598-021-88802-7
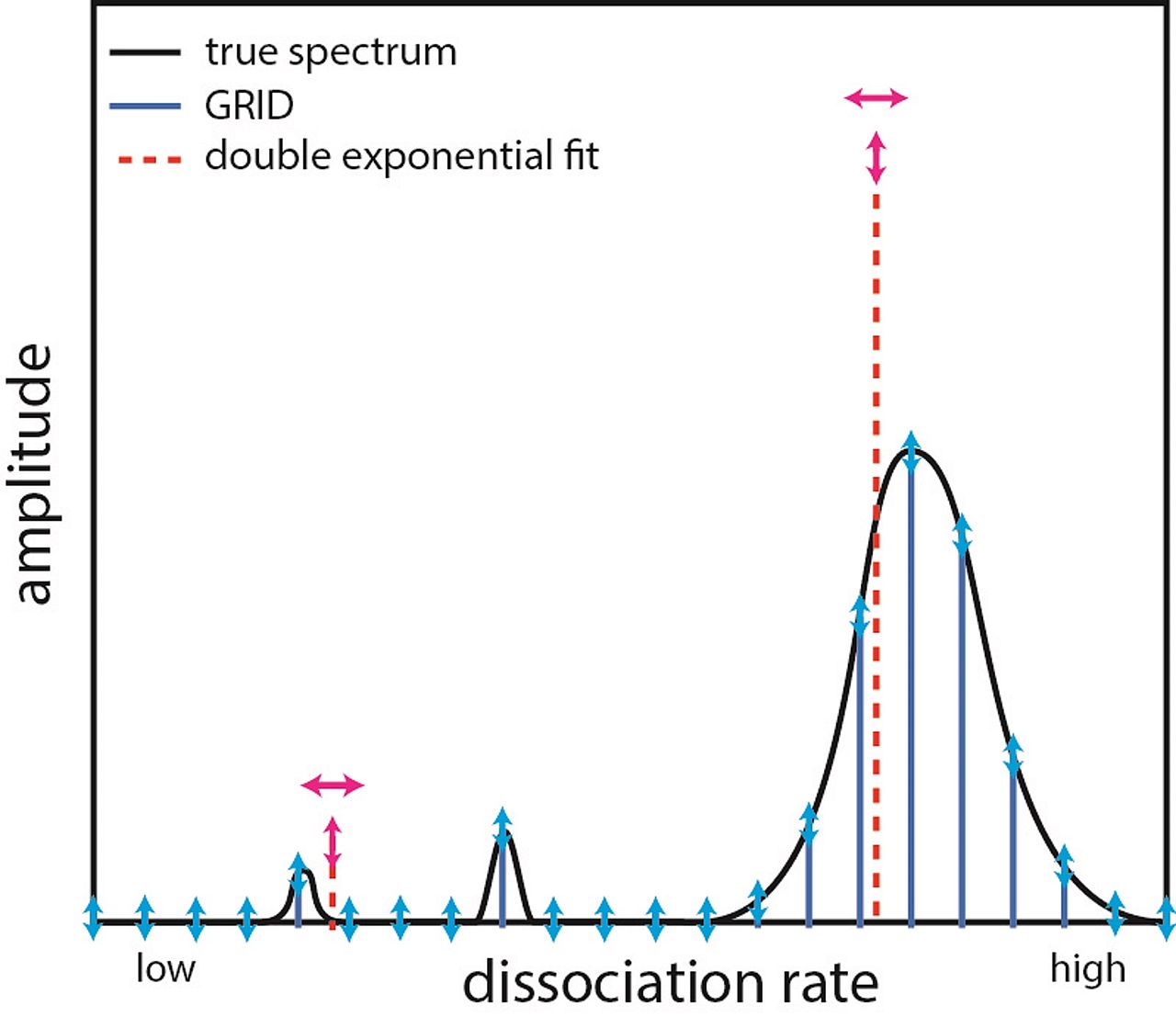
Inferring quantity and qualities of superimposed reaction rates in single molecule survival time distributions
Actions of molecular species, for example binding of transcription factors to chromatin, are intrinsically stochastic and may comprise several mutually exclusive pathways. Inverse Laplace transformation in principle resolves the rate constants and frequencies of superimposed reaction processes, however current approaches are challenged by single molecule fluorescence time series prone to photobleaching. Here, we present a genuine rate identification method (GRID) that infers the quantity, rates and frequencies of dissociation processes from single molecule fluorescence survival time distributions using a dense grid of possible decay rates. In particular, GRID is able to resolve broad clusters of rate constants not accessible to common models of one to three exponential decay rates. We validate GRID by simulations and apply it to the problem of in-vivo TF-DNA dissociation, which recently gained interest due to novel single molecule imaging technologies. We consider dissociation of the transcription factor CDX2 from chromatin. GRID resolves distinct, decay rates and identifies residence time classes overlooked by other methods. We confirm that such sparsely distributed decay rates are compatible with common models of TF sliding on DNA.
https://www.biorxiv.org/content/10.1101/679258v1
Software https://gitlab.com/GebhardtLab/GRID