Mechanismen genetischer Epilepsien
Die Epilepsien gehören zu den häufigsten Erkrankungen des zentralen Nervensystems, die ca. 3% der Bevölkerung im Laufe des Lebens betreffen. Bis zu 50% aller Epilepsien sind vorwiegend genetisch bedingt, insbesondere die idiopathischen Epilepsien, bei denen sich keine Hinweise auf eine äußere Ursache oder auf Veränderungen der Struktur des Gehirns finden. Epileptische Anfälle entstehen durch spontane, unkontrollierte Entladungen einer Gruppe von Nervenzellen oder auch des gesamten Gehirns. Diese Entladungen kommen durch eine vermehrte Erregbarkeit zustande. Genetisch bedingte Funktionsstörungen von Ionenkanälen spielen eine zentrale Rolle bei der Entstehung von idiopathischen Epilepsien. Dies ist gut verständlich, da Ionenkanäle, die den Ionenfluss durch die Zellmembran regulieren, die Basis für die Erregbarkeit aller Nervenzellen darstellen. Deshalb wirken auch Medikamente, die zur Behandlung von Epilepsien eingesetzt werden, ganz überwiegend auf Ionenkanäle.
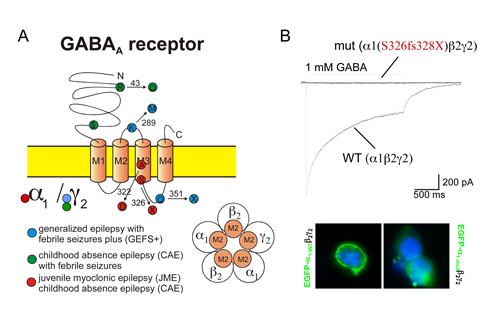
Abb. 1. Funktionsverlust des GABAA Rezeptors bei idiopathischen Epilepsien.
(A) Schematische Darstellung des GABAA Rezeptors in der Zellmembran. Dies ist der wichtigste Rezeptor für hemmende Einflüsse auf Nervenzellen im Gehirn. Er setzt sich aus fünf ähnlichen Untereinheiten zusammen. Die häufigste im Gehirn vorkommende Form besteht aus zwei α1-, zwei β2- und einer γ2-Untereinheit (unten rechts). Mutationen wurden bisher in der α1-Untereinheit oder in der γ2-Untereinheit bei den genannten Epilepsiesyndromen gefunden. Symbole: Die Buchstaben stehen für Aminosäuren, X für ein Stopcodon, $ für eine mutierte Spleißstelle. Für alle Mutationen konnte gezeigt werden, dass sie zu einem partiellen Funktionsverlust des GABAA Rezeptors führen. Wenn die Hemmung nicht mehr effektiv funktioniert, können epileptische Anfälle entstehen.
(B) Beispiel für einen kompletten Funktionsverlust des GABAA Rezeptors für die Mutation S326fs328X, die zu einem verkürzten Protein führt. Während für den Wildtyp Rezeptor (WT) durch Applikation von 1 mM GABA (γ-amino-Buttersäure) ein rasch aktivierender und dann desensitisierender Membranstrom ausgelöst werden kann, erhält man für den mutierten Rezeptor gar keinen Strom (oberer Teil). Dies ist durch einen fehlenden Transport des mutierten Rezeptors zur Zellmembran bedingt (unten): Man sieht eine bandförmige grüne Anfärbung der Oberflächenmembran transfizierter Zellen für den WT (links), während diese für die Mutation gänzlich ausbleibt (rechts). Abb. modifiziert nach der Arbeit: Maljevic S, Krampfl K, Cobilanschi J, Tilgen N, Beyer S, Weber YG, Schlesinger F, Ursu D, Melzer W, Cossette P, Bufler J, Lerche H, Heils A. A mutation in the the GABAA receptor ±1-subunit is associated with absence epilepsy. Ann Neurol 2006;59:983-7
Die aktuellen Kenntnisse zeigen zwar eine genetische Heterogenität, jedoch finden sich konkrete Hinweise auf eine funktionelle Konvergenz. So scheinen die Einflüsse hemmender Nervenzellen, die den Botenstoff γ-amino-Buttersäure (GABA) zur Signalübertragung verwenden (sog. GABAerge Interneurone), besonders häufig betroffen zu sein. Einerseits kann die eigene Erregbarkeit dieser GABAergen Nervenzellen vermindert sein, weil ihre Natriumkanäle nicht mehr richtig funktionieren und sie deshalb weniger Impulse an andere Nervenzellen weiterleiten können. Andererseits kann auch die Übertragung ihrer hemmenden Einflüsse auf die erregenden Nervenzellen gestört sein, da die Rezeptoren für den hemmenden Botenstoff GABA funktionsuntüchtig sind (s. Abb. 1). Durch die in beiden Fällen verminderte Hemmung kommt es zu einer Übererregbarkeit der Nervenzellen und es können epileptische Anfälle entstehen. In einem weiteren Beispiel kann neuronale Übererregbarkeit entstehen, wenn das Gleichgewicht von Strömen durch Natrium- und Kaliumkanäle an einer Schlüsselstelle der erregenden Nervenzellen gestört wird. An dieser Stelle entstehen die weiterzuleitenden Impulse (Aktionspotenziale) und deren Frequenz (Feuerungsrate) wird reguliert. Ein vermehrter Natriumstrom oder ein verminderter Kaliumstrom führen zu einer Übererregbarkeit, die epileptische Anfälle auslösen kann.
Durch Untersuchungen der genetischen Ursachen und der resultierenden Krankheitsmechanismen kann die Entstehung von Epilepsien besser verstanden und ihre Therapie verbessert werden. So können sich neue Zielstrukturen für die Therapie mit Medikamenten ergeben, die bisher nicht genutzt werden. Eine neue Substanz, die zu einer vermehrten Aktivität von Kaliumkanälen führt, und damit das Gegenteil von krankheitsverursachenden Mutationen bewirkt, befindet sich derzeit in klinischer Erprobung.
Kontakt
- Prof. Dr. med. Holger Lerche
- Neurologische Klinik und Institut Angewandte Physiologie
Neurologische Klinik im RKU- Oberer Eselsberg 45
- D-89081 Ulm, Germany